
Background
G-CSF (Granulocyte Colony-Stimulating Factor), together with GM-CSF (granulocyte-macrophage colony stimulating factor) and M-CSF (macrophage colony stimulating factor), are members of the CSF (colony stimulating factor) family. This family of growth factors (or cytokines) was first described for its ability to stimulate in vitro the proliferation of immune cells such as granulocytes and macrophages [1]. Granulocytes are composed of four cell types of the immune system: Basophils, Eosinophils, Neutrophils and Mast cells. They are produced in the bone marrow where they undergo differentiation in their subtypes and go to the bloodstream (with the exception of the mast cells), where they will constitute most of the so-called white blood cells that are responsible for the immune defense of our body [1,2]. G-CSF binds to the surface of responsive cells through a homo-dimeric transmembrane receptor designated G-CSF-R (also known as CD114). This binding causes activation of the intracellular domain of the receptor with a kinase activity capable of phosphorylating and activating various intracellular signals such as the Jak-Stat (Janus kinase - Signal Transducer and Activator of Transcription protein), MapK1/2 (mito-gen-activated protein kinase 1 and 2) and PI3K (phosphatidylinositol 3-kinase) family [2,3]. The family activated will depend on the cell type and physiological condition of the responsive cells, which can be bone marrow cells, various types of leukocytes, cancer cells and nerve cells, among others. Activation of the intracellular signaling cascades by G-CSF provokes, among other effects, the stimulation for cell proliferation [3]. Leukopenia is an anomalous state where is a small number of circulating white blood cells, which can compromise the immune defenses of the patient with this condition. Leukopenia can be caused by a number of factors: genetic abnormality, bone marrow transplant, some types of cancer, treatment with radiation therapy and chemotherapy, viral infections, among other conditions [2]. One of the alternatives for treating leukopenia is to increase circulating granulocyte levels using purified G-CSF in biopharmaceutical applications [4]. Due to its applications in medicine, human G-CSF (human Granulocyte Colony-Stimulating Factor, hG-CSF) has a high added value, with an annual $5 billion market only in the United States, which make it an interesting candidate to be produced in bioreactors [2,5]. G-CSF, as well as any biologically complex molecule expressed by mammals, undergoes post-translational modifications that may change their biological activity in either prokaryotic (bacteria) or simple eukaryotic (fungi and yeast) production systems. Therefore, the production of its most human-similar form is frequently achieved in genetic modified mammalian-cell culture system in vitro [6]. However, this platform for production of biomolecules is of high cost and low efficiency, and more efficient production alternatives such as the mammary gland have been advocated in recent years by various research groups and institutes around the world [5,7]. Therefore, the production of biomolecules of biotechnological interest in animal models is of high biotechnological interest and if the platform is a dairy animal, it is of interest to the agricultural sector as well. In this study, we aimed the production of transgenic hG-CSF bovine through the lipofection of bovine primary fibroblasts with an hG-CSF expression cassette and cloning these fibroblasts by the somatic cell nuclear transfer (SCNT) technique.
Material and Methods
All reagents used in this study are purchased from Sigma-Aldrich (São Paulo, Brazil), unless specified.
The cassette with the β-Casein (bovine) promoter and the hG-CSF gene was obtained by digestion of the vector pCR2.1-hG-GSF, kindly provided by the RDA (Rural Development Agency, South Korea), with the enzyme Not I. The electrophoresis of the products of this digestion produced two DNA fragments (4.8 kb of the G-CSF cassette and 5.5 kb of the pCR2.1 vector, Invitrogen). The 4.8 kb fragment were purified from the gel using the Wizard SV Kit (Promega, Fitch-burg, WI, USA). The G-CSF cassette was cloned into the Not I site of the pCiNeo vector (Promega) by the enzyme T4 DNA Ligase (Promega), following the manufacturer’s recommendations (supplementary Fig. S1). The binding system was transformed into E. coli DH5 α by thermal-shock transformation [8]. Ampicillin-resistant (100 η g/mL) colonies had their plasmid DNA extracted by the salting-out method [9]. The isolated clones were confirmed by digestion with the Eco RI enzyme (Promega) which has the ability to discriminate the pCiNeo with the insert and its correct orientation. The final construct (pCiNeo-G-CSF) was confirmed by DNA sequencing (Helixxa Genomic Services S.A., São Paulo Brazil).
Fibroblasts were isolated from a biopsy obtained from the skin of a four years old female Holstein and cultured in vitro according to protocols already established in the Laboratory of Animal Reproduction of Embrapa Genetic Resources and Bio-technology [10]. The isolated cells were cultured in DMEM medium (Gibco BRL, Billings, MT, USA) enriched with 10% fetal bovine serum (FBS) to the 2nd cell pass and frozen in culture medium containing 10% Dime-thylsulfoxide (Sigma, Marlborough, MA, USA), in 0.25 mL vials and stored in liquid nitrogen. Such stock cells were used throughout this work to provide the fibroblasts for insertion of the genes of interest and thereafter as nucleus donors in the TNSC procedures.
Bovine fibroblasts of primary origin, obtained from LRA from skin biopsies, were cultured in vitro (with D-MEM medium + 10% FBS) at 38°C in humidified incubator. These cells were transfected with the DNA fragment containing the hG-CSF cassette and the Neo gene, removed from the pCiNeo-G-CSF vector with Sac I and Bam HI enzymes (Promega). The 6,800 bp fragment containing the G-CSF cassette and the Neomycin/G418 resistance gene (NeoR) was purified from agarose gel (0.8%) with Wizard-SV Kit (Promega), quantified by spectrophotometry and confirmed by agarose gel staining. On the day prior to transfection, 200,000 to 250,000 fibroblasts were seeded into 24 well plates and allowed to adhere to the bottom so that the next day they reached 60%–80% confluence of the well area. In transfections, 0.25 η g of the pEF-GFP (green fluorescent protein) vector (Addgene, Cambridge, Massachusetts, USA) expressing the GFP was used as a positive control. The DNAs were mixed with 100 μL of serum-free D-MEM (Dulbecco Modified Eagle Medium) and subsequently added with 1 μL of Plus reagent (Invitrogen). The DNA/Plus reagent complexes were incubated at room temperature for 5 min. After incubation, 100 μL of D-MEM (without fetal bovine serum-FBS) containing 1.5 μL of Lipofectamine LTX reagent (Invitrogen) was added to the DNA/Plus complex. The DNA/Plus/TX complex was incubated at room temperature for 20 min. Cells were transfected with the DNA/Plus/LTX complex for 6 hours at 38°C. After transfection the medium was replaced by D-MEM + 10% FBS medium and cultured for more 24 h at 38°C. After this time, the medium was replaced with D-MEM + 10% FBS with the antibiotic G418 (Invitrogen) at 0.8 mg/mL. After 8 to 10 days, all cells lacking the integrated NeoR gene in their genome were killed by the G418. As a negative control, we used non-transfected fibroblasts exposed to G418. At two weeks the screening process was completed and the cells made available to verify the presence of the G-CSF/NeoR cassette in their DNA, as well as to be used as donors of DNA in TNCS. The presence of G-CSF/NeoR in the genome of the transfected cells was investigated by PCR, with specific primers, as described before [10].
The bovine ovaries used were collected at slaughterhouse and the follicles measuring from 3 to 8 mm were aspirated with the aid of a 19G needle connected to a vacuum system. The oocytes collected were classified based on the cumulus cell layer and homogeneity of the cytoplasm, and placed in 200 μL drops by 18 to 22 h in in vitro maturation medium, consisting of 199 medium (Invitrogen), supplemented with 10% FBS (Invitrogen), 10 μg/mL FSH (follicle stimulating hormone), 100 IU/mL penicillin and 50 μg/mL streptomycin, covered with mineral oil and kept in incubator at 39°C, 5% CO2 and saturated humidity.
The SCNT employed in our study was performed as described in the literature [11], with some modifications described previously by our group [12]. After the maturation period, the oocytes were denuded in 0.2% hyaluronidase and selected according to the expulsion of the first polar corpuscle and cytoplasmic quality. Before enucleation (removal of nuclear DNA) the oocytes were incubated for 20 minutes with 2 μg/mL DNA dye Hoechst 33342 and 3.33 μg/mL cytochalasin D, diluted in maturation medium. The enucleations were performed under a Nikon inverted microscope (Nikon, Tokyo, Japan), with the aid of a TransferMAN NK2 electronic micromanipulator (Eppendorf, Hamburg, Germany). The process involves the removal of a small portion of cytoplasmic material accompanied by the metaphase plate adjacent to the first polar corpuscle, confirmed by visualization under UV light of the DNA stained with Hoechst 33342. During reconstruction, the donor cells of the genomic DNA (transfgenic fibroblasts) were inserted into the perivitelline space. The karyoplast complexes were positioned between two parallel electrodes with fusion medium and received DC pulses of 2.5 kV/cm, generated by the BTX-ECM 200 electrofusion apparatus (Harvard Apparatus, Inc., Holliston, MA, USA). After fusion, the karyoplast complexes were artificially activated using 5 μM Ionomycin prepared in SOFaaci [13] with addition of FBS followed by incubation with 2 mM 6-dimethyl-aminopurine (6-DMAP) prepared in synthetic oviductal fluid (SOF) with addition of FBS.
The reconstructed karyoplast complexes were co-cultured in 9.2 cm2 Petri dishes (Corning, Midland, Michigan, USA) over a layer of bovine granulosa cells in 160 μL drops of SOFaaci medium supplemented with 5% FBS, covered with silicone oil, and kept in humidified incubators at 39°C, 5% CO2 and saturated humidity. The development rate were checked on day 2 for evaluation of cleavage, and day 7 for evaluation of blastocyst rate. Nellore heifers were used as recipients for the SCNT embryos. Prior to the embryo transfer, they were submitted to a fixed-time embryo transfer synchronization protocol, which consists of placement of an intra-vaginal implant containing 1 g of progesterone (day 0 –D0) and an i.m. injection of 2 mg of estradiol benzoate in D5, 150 μg of PGF2α (prostaglandin alfa) was i.m. injected in D8, the progesterone implant was withdrawn in D9 and 1 mg of estradiol benzoate was injected before blastocyst embryos were transferred. On the day of the transfer the recipients were evaluated for location and classification of the corpus luteum size as class I, II, III (large, medium and small respectively). In those who were able to receive the embryo (class I and II), epidural anesthesia was performed with 3 mL of 2% lidocaine hydrochloride in the sacro-coccix space to relax the pelvic region. For anovulation, an applicator was used by inserting the vial containing the embryo and depositing the embryos in the ipsilateral horn to the corpus luteum. After 30, 60, and 90 days after embryo transfer, transverse rectal ultrasonography (Aloka SSD-500, Overseas Monitor Corporation, Richmond, BC, USA) with a 7.5 MHz linear transducer as used to evaluate pregnancy and follow-up the fetal development. Rectal palpation was also used to assess the texture of the placenta, as well as the number and distribution of the placentomes.
Embryo vitrification was performed as previously described by [14], with minor modifications. A holding medium (HM) was used to handle embryos during vitrification and warming procedures, and was composed of HEPES buffered TCM-199 (Invitrogen®) supplemented with 20% FBS. For vitrification, the groups of embryos were first washed three times in an equilibrium solution composed of 7.5% ethylene glycol (EG) and 7.5% dimethyl sulfoxide (ME2SO) dissolved in HM for a minimum of 9 minutes. Embryos were then transferred to a vitrification solution consisting of 15% EG, 15% ME2SO, and 0.5 M sucrose in HM, and incubated for 45 to 60 seconds. Next, embryos were placed in a Cryotop® device (Kitazato Corp., Japan) in sets of 3 to 5 embryos under a stereomicroscope. Subsequently, the Cryotop® was immediately submerged in liquid nitrogen. The vitrified embryos were stored in the liquid nitrogen container, until an adequate moment for transfer into hormone-synchronized female recipients. At that time, warming was performed by immersing the end of the Cryotop® for 1 minute in a drop of HM supplemented with 1 M sucrose that had been pre-warmed to 37°C. The embryos were transferred to HM supplemented with 0.5 M sucrose for 3 minutes and then finally transferred to original HM. Subsequently, the warmed embryos were cultured for an additional 4 hours on SOF medium. At the end of this period, the embryos were evaluated for re-expansion and developmental stage progression rates. All embryos presenting no morphologically degenerating signals were considered as having survived. Embryos that changed their developmental stage during the culture period between vitrification and post warming evaluation were transferred.
Bovine skin fibroblasts isolated as described above were cultured in D-MEM + 10% FBS medium (Gibco, BRL) and collected at different times of cell growth. This separation was done according to the confluence state, measured in percentage of surface occupation of the cell-culture bottle, as well as the time elapsed after reaching 100% confluence (100% surface occupation by fibroblasts). After collection, the fibroblasts were fixed in 70% ethanol (Sigma) and stained with 500 μg/mL Propionic Iodide (Sigma) solution as described [15]. The amount of DNA / cell content and cell cycle stage classification analyzed by cytometry in Flow Sight cytometer (Amnis-Merck KGaA).
The genetic identity of the cells and animals used in this work: fibroblast of nucleus-donor animal, receptor heifer and transgenic fetus clone was determined by genetic identity examination performed with 16 microsatellite probes certified by ISAG (International Society for Animal Genetics) for cattle. The genotyping and genetic identity report of the DNA samples was issued by Geneal Diagnósticos LTDA. The confirmation of transgene cassette insertion was done by PCR using the primers: G-CSF Exon 4 Foward (5’-CCCACCTTGGACACACTGCAGCTGG), G-CSF Exon 5 Reverse (5’-TCAGGGCTCAGCAAGGTAGCG) and pCiNeo Reverse (5’-GACAAGCTTACACTTTATGCTTCCGGC). PCR (polymerase chain reaction) conditions: 1.25 mM MgCl2; 0.4 mM dNTP mix; 10 ρmoles of primers and 1.5 units of Taq Polymerase (Invitrogen). Thermal cycling: 95°C / 1 min; 40× (94°C / 20 sec; 65°C / 20 sec; 72°C / 20 sec); 72°C / 5 min; 12°C / ∞.
Results
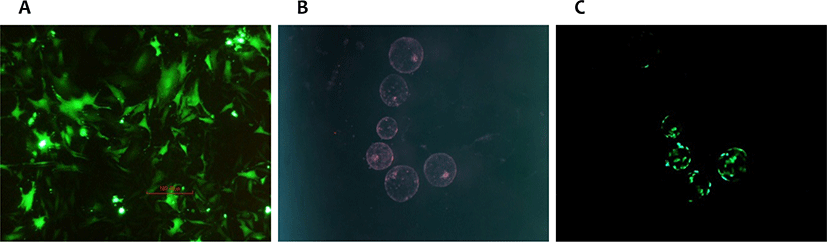
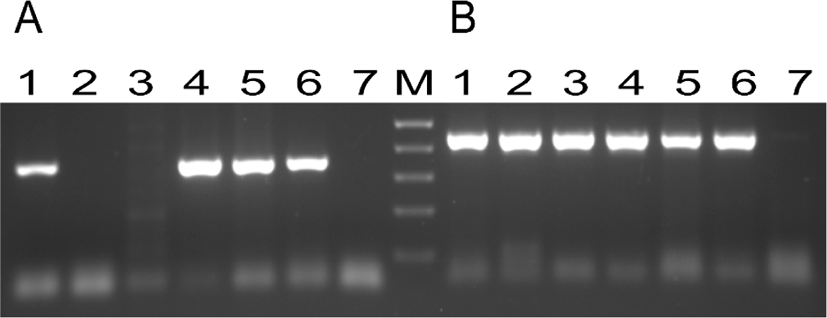
The transfection of bovine fibroblasts by lipofection was successful, as can be seen in the positive control expressing the GFP, transfected with the LTX reagent and the pEF-GFP vector (Fig. 1A). Transfection with the DNA fragment containing the G-CSF and NeoR expression cassettes was also successful due to the selection response to the antibiotic G418, which eliminated 100% of the untransfected cells in 14 days (data not shown). The DNA of G418 selected cells, containing the hG-CSF/NeoR expression cassette, was submitted to PCR amplification with primers specific for the inserted cassette, confirming the integration of the cassette into the fibroblast genome (Fig. 2, line 1).
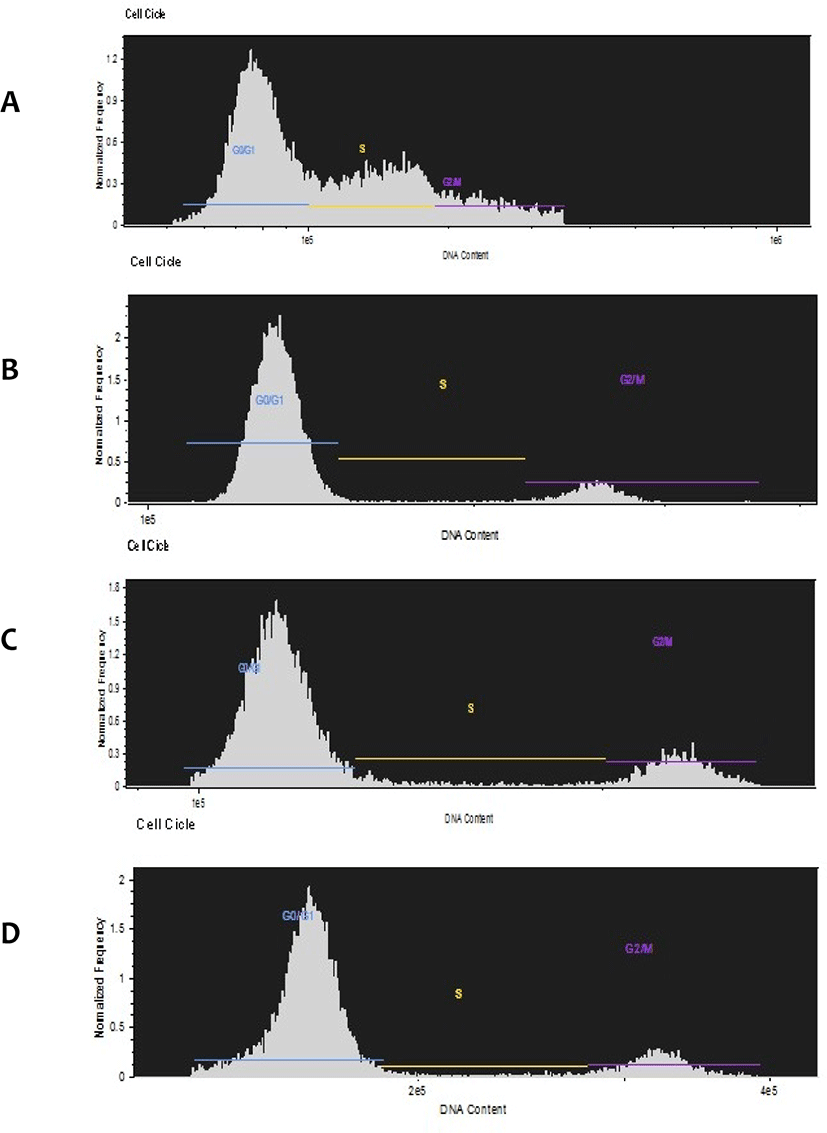
To confirm that the cells donor used in our SCNT experiments were correctly synchronized in G0/G1, we collected growing cells (60% confluence) and stationary growth phase cells (100% confluence) at different times (0 h, 60 h, 120 h). The collected cells were fixed and stained with Hoechst 33342 and analyzed by flow cytometry to separate the G0/G1 (2N, diploid genome content), S (DNA synthesis) and G2/M (mitosis, 4N genome content) populations. The results indicated that growth inhibition by contact and consequent synchronization of the G0/G1 cell population was efficiently achieved, even within the first few hours after confluence observation (Fig. 3), indicating that the vast majority of cells used in SCNT were at the correct cell cycle stage advocated for a successful SCNT cloning event.
To test our capacity of producing TG-SCNT embryos, bovine fibroblasts transfected with an enhanced GFP (eGFP) cassette, but not selected with G418, were used as a nucleus donor in SCNT procedure. This pilot test produced blastocysts expressing the eGFP reporter gene or not, depending on the incorporation of this cassette into the genome of nucleus donor cells (Fig. 1B and 1C). In a second SCNT assay, the bovine fibroblasts transfected with hG-CSF/NeoR cassette and selected with G418 were used for the reconstruction of karyoplasts, producing 103 embryos, which were transferred to 79 recipient heifers, generating two pregnancies (Table 1). The fetus of the first pregnancy was detected by ultrasonography 30 days after the TG blastocyst anovulation, but it was no longer detected in the subsequent ultrasonography at the 60th day of gestation. The second pregnancy developed until the seventh month of gestation, when a spontaneous abortion of the cloned TG fetus occurred. During this gestation, the ultrasonography and rectal palpation examination, conducted monthly, showed an anomalous number and distribution of placentomes, indicating a negative prognosis for this gestation. After the spontaneous abortion, genomic DNA was extracted from the aborted fetus tissues: mammary gland, skin and muscle, as well from blood samples of the fibroblasts of the donor cow and the recipient heifer of the TG-clone fetus. The presence of the hG-CSF/NeoR cassette was detected by PCR only in samples from the TG fetus and TG cell line, as expected (Fig. 2). The genetic identification of the DNA samples was done using 16 single sequence repeat (SSR) probes, and the identity between the nucleus donor animal and the aborted fetus was confirmed (Table 2).
| Embryo’s type | Activated structures | Blastocyst rate (%) | Transferred embryos | Embryo recipients | Pregnancy detected |
|---|---|---|---|---|---|
| Paternogenetic control | 743 | 34.8 | 259NT | - | - |
| Fresh | 529 | 17.2 | 91 | 69 | 2 |
| Vitrified | 204 | 19.61 | 292 | 18 | 0 |
| Total (Fresh+vitrified) | 733 | 17.9 | 120 | 873 | 24 |
40 Blastocysts were vitrified, stored in N2 and thawed. Twenty-nine of them (72.5%) presented re-expansion and progress in post thawing development.
The recipient heifers were reused several times, respecting a minimum interval of one month between successive synchronizations.
Discussion
The cells expressing eGFP were used to test the SCNT parameters and protocol adopted in our laboratory previously [12], producing GFP expressing blastocysts as expected (Fig. 1B and C). Since in this test with GFP vector the G418 selection was not used, some SCNT blastocysts did not express GFP, or expressed GFP in a mosaic pattern as expected. However, with this pilot test we could proceed with the SCNT cloning using the cells transfected with the hG-CSF/NeoR cassette and selected with G418. The literature data on SCNT cloning point to the importance of the cell cycle phase during the reconstruction of embryo clones in the correct process of genome reprogramming. The correct cell-cycle synchronism between the nucleus donor cell and the MII oocyte used in the SCNT is of pivotal importance for the development of a competent clone embryo, with full potential to become a fetus that allows the birth of a healthy animal [11,16,21]. Failures in this process result in clones that are born weak and do not survive the first month of life. Considering this information, in order to be able to have a chance of successful cloning, the prerogative is that the donor nucleus cell be in the G1/G0 phase of the cell cycle [16]. Synchronization of the fibroblast culture in the G1/G0 phase (with stoppage in the cell division) can be obtained by several methods; among them serum deprivation and the inhibition of cell division by contact (confluence of 100%) are frequently used [18]. In our laboratory, we used contact inhibition as a G0/G1 synchronization method, which was shown to be an effective method to synchronize the nucleus donor cells as demonstrated by the cell cycle analysis of the confluent fibroblast cell culture. In this study, we produced 103 SNTC embryos at the blastocyst stage, 80 of them were transferred freshly to 61 recipient cows, producing two pregnancies. However, due to the low pregnancy rate observed during the dry season anovulations, probably due to the worse physical status of the recipient heifers, we decided to freeze by vitrification all GM embryos produced in the second dry season and to transfer them together in the middle of raining season, when the recipient corporal score was imporved. This strategy aimed to optimize the synchronization of the recipient cows during a season with more appropriate weather condition, even knowing that the vitrification would reduce the quality and viability of the transferred embryos. This approach produced 31 expanded embryos, which, after thawing, 23 were transferred to 18 recipients (mostly transferred in pairs). This approach did not produce any positive pregnancies after 30 days of the anovulation and was no longer used later in this study. From the two pregnancies produced by the freshly transferred SCNT blastocysts, one stopped its development between the 30th and 60th day of gestation, being reabsorbed probably due to defects in the fetus development provoked by an inefficient genome reprogramming [17,19,20]. The second pregnancy developed until the 7th month of gestation, when an abortion of the fetus happened naturally. The fetus was collected as soon as it was found in the pasture, but it was in an advanced stage of decomposition. Besides its decomposition stage, we could collect samples from three tissues: mammary gland, skin and muscle. The genomic DNA was extracted from these samples and used to confirm the presence of the hG-CSF/NeoR cassette, and to confirm the genetic identity between the TG-fetus and the transfected fibroblast cell line used as the nucleus donor during the SCNT, as well as the identity with the cow from whom these fibroblasts were obtained. During the gestation of the TG fetus, the ultrasound and rectal palpation examinations showed a placenta with less placentomes than expected and with an abnormal distribution of them. This malformation of the placenta is frequently observed in SCNT-clones pregnancies, and is correlated with an abnormal or inefficient genetic reprogramming of the genome of nucleus donor cells [19,20]. These problems observed in SCNT-clone placentation is one of the major causes of the high fetal losses and low efficiency intrinsic of SCNT technique [22].
Conclusion
Although this project did not reach its initial objective of delivering live transgenic cattle expressing the hG-CSF, the knowledge produced by this work allows us to keep following to attempt the production of transgenic cattle expressing valuable biomolecules, which is so promising and strategic for Brazil.