
INTRODUCTION
Lumpy skin disease (LSD) is one of the most important animal poxviruses because of its serious economic consequences on the agricultural industry. LSD is characterized by fever, swelling of peripheral lymph nodes, reduced milk production, and skin nodules. The disease causes high morbidity and low mortality rates [1]. LSD is caused by a virus (LSDV) in the family Poxviridae, genus Capripoxvirus. During an outbreak, large-scale immunization of bovines is the most effective control measure when combined with bovine movement restrictions. Nowadays, the majority of commercially available vaccines against LSD are live attenuated vaccines (LAVs) based on LSDV, sheeppox virus (SPPV), or goatpox virus (GTPV) [2]. Many LAVs, however, demonstrate virulence reversal by back-mutation, recombination, reassortment, or change of quasispecies diversity [3].
In recent years, there have been reports of LSDV outbreaks in vaccinated areas, indicating the possible emergence of mutated strains of the virus [4,5]. This is concerning because these new variants may become more virulent or may not be effectively controlled by existing vaccines, leading to more outbreaks and potential economic losses for the livestock industry [6,7]. In Thailand, LSDV recombinant strains were found in several regions during the first outbreak in March 2021 [8–11]. LSDV72/Prachuap Khiri Khan / Thailand / 2021 and LSDV / Thailand / Yasothon / 2021 sequences showed the highest identity to the Chinese and Vietnamese strains [8,12]. Since June 2021, LAVs from Lumpyvax (MSD, Midrand, South Africa) and Kemin (MEVAC, Cairo, Egypt) have been used to prevent their spread. Therefore, ongoing surveillance and research are needed to understand the evolution of LSDV in response to vaccination as well as develop new strategies for controlling the disease. This study aimed to monitor the changes in the genetic characteristics of LSDV after vaccination in Thailand. This information will be useful for developing effective diagnosis and control strategies to prevent and manage future LSDV outbreaks.
MATERIALS AND METHODS
Interviews with cattle owners were conducted following the guiding principles for the care and use of research animals. The protocol was approved by the KASETSART UNIVERSITY Institutional Animal Care and Use Committee (Project approval number ACKU64-VET-070). Animal information was gathered with permission from the owners.
Five LSDV DNA samples with Ct values less than 20 and animal histories were collected from government laboratories under the Department of Livestock Development (DLD); four samples were kindly provided by the Veterinary Research and Development Centers (VRDCs) in the North, Upper Northeast, West, and Upper South regions, and one sample was provided by the National Institute of Animal Health (NIAH). All samples came from non-vaccinated cattle with LSD clinical symptoms that were living in vaccinated areas.
A 20 µL-reaction mixture containing 200 nM primer, 120 nM probe, 5 µL of DNA template, and 10 µL of FastStart Essential DNA Probes Master (Roche, Indianapolis IN, USA) was used to conduct real-time PCR for the initial screening of an LSD-positive case. CaPV-074F1 5′-AAA ACG GTA TAT GGA ATA GAG TTG GAA-3′ and CaPV-074R1 5′-AAA TGA AAC CAA TGG ATG GGA TA-3′ were used with the minor groove binder (MGB) and the TaqMan probe CaPV-074P1 5′-FAM-TGG CTC ATA GAT TTC CT-MGBNFQ-3′ [13]. The following thermal cycler conditions were set up in a QuantStudio 5 (Thermo Fisher Scientific, Waltham, MA, USA): A 10-minute denaturation process at 95°C was followed by 40 cycles of amplification (15 s at 95°C and 45 s at 60°C). The LSDV DNA levels were represented as threshold cycle (Ct) values.
The Nextera XT DNA library preparation kit was used to build the DNA library. The sequencing was done using a MiSeq benchtop sequencer (Illumina, San Diego, CA, USA) with a MiSeq reagent kit version 3 and 2 × 300-bp paired-end sequencing. Using the FastQC software (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/), raw data quality was evaluated. (Accessed on December 18, 2022). Geneious Prime software, version 2021.2.2 (Biomatters, Auckland, New Zealand), was used for genome assembly and annotation. A BBDuk Trimmer was used to trim the readings based on length (>20 bp) and quality (Q score > 30). Using the SPAdes assembler version 3.15.2, the trimmed reads were de novo assembled into contigs and aligned to the LSDV/KM/Taiwan/2020 (OL752713) and LSDV/Thailand/Yasothon/2021 (OM033705) reference genomes.
MAFFT alignment in Geneious Prime software was used for aligning the research sequences with CaPV strains from GenBank [14]. The whole genome sequencing (WGS) phylogenetic tree was built using the Neighbor-Joining method and 1,000 bootstraps. The genetic distances were calculated in Geneious Tree Builder using the Tamura-Nei model with the default settings. The sequence was aligned with LSDV/Thailand/Yasothon/2021 (Yasothon strain) using MAFFT pairwise alignment in Geneious Prime software to evaluate mutation. Using the program, the annotation was transferred from the Yasothon strain. The discover variations/single nucleotide polymorphisms (SNPs) tool was used to locate the SNPs. The translation function indicated amino acid alterations. The depth of coverage for all mutation locations was documented to guarantee that the mutation did not result from human or technical errors during analysis.
RESULTS
All samples were collected after the implementation of vaccination campaigns in the sample areas. The history of five animals recorded on the sample submission forms indicated that skin lesions were collected from cattle that exhibited clinical signs of LSD between October 2021 and December 2022. These five animals came from five different regions of Thailand including the North (Chiang Mai), Northeast (Khon Kaen), Central (Nakhon Pathom), West (Prachuap Khiri Khan), and South (Trang). Notably, there was no sample from the East region. The Ct values of LSDV p32 from all five samples were less than 20.
The phylogenetic tree showed that LSDV could be clustered into three groups including filed, vaccine, and recombinant strains. All Thai LSDVs were classified into recombinant strains that were closely related to the Vietnamese strain (Fig. 1). The total number of reads, depth of coverage, and nucleotide identities compared to the Yasothon strain are presented in Table 1. The full genome sequences of five LSDVs were deposited in the GenBank database to obtain accession numbers (Table 1). The length of whole genome sequences ranged between 150,653 and 150,812 nucleotides with 156 predicted protein-coding genes.
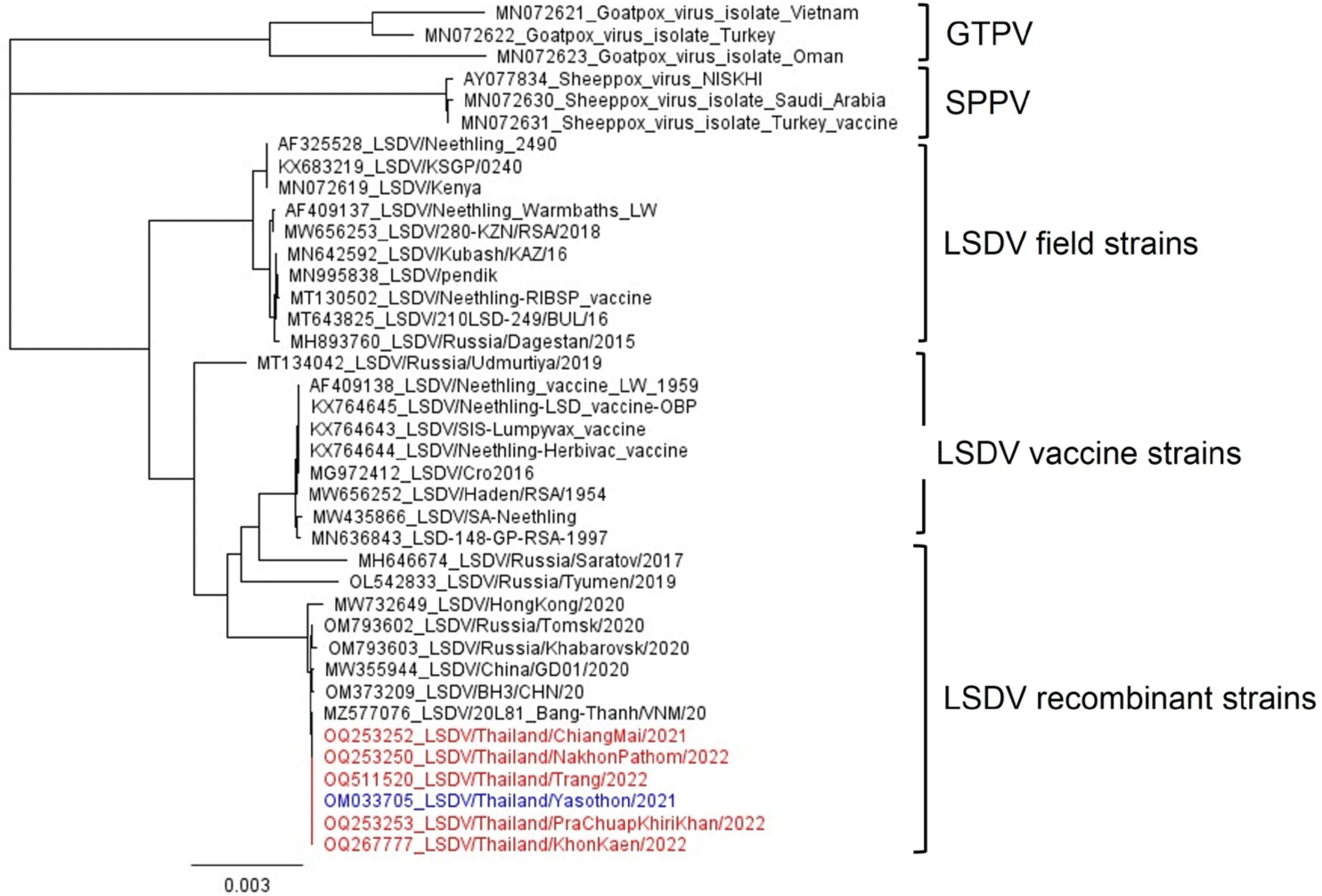
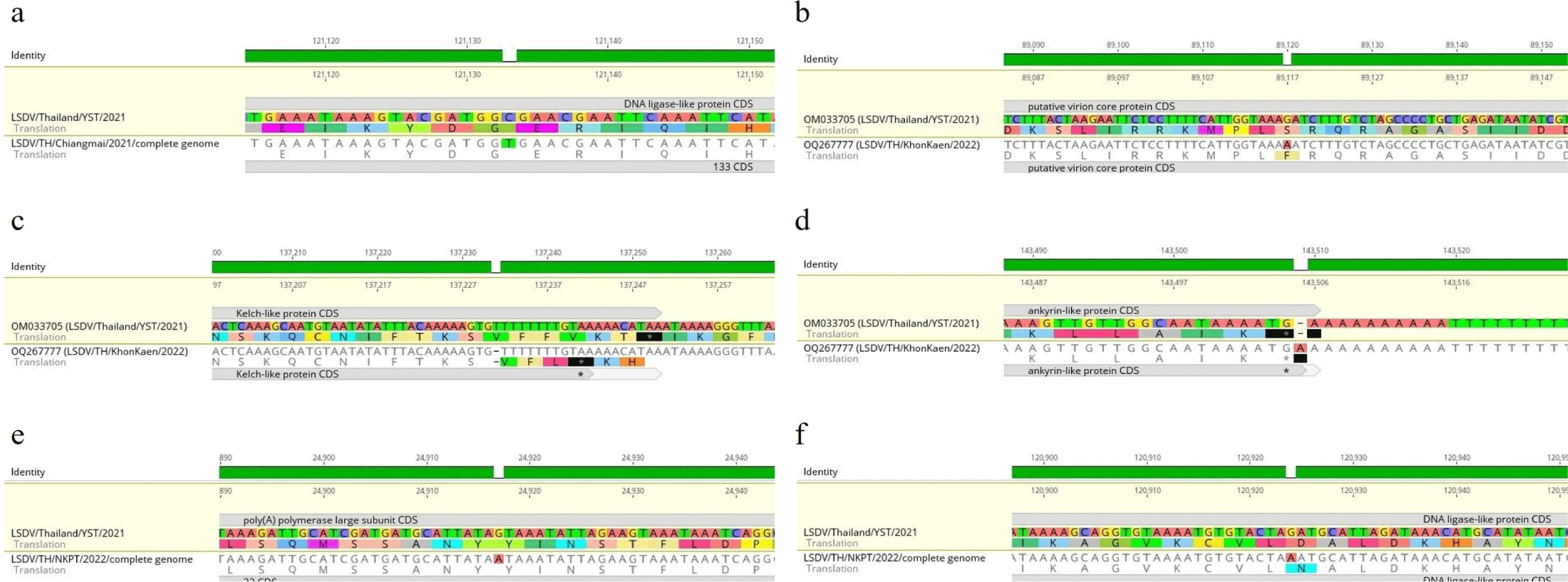
Compared to the Yasothon strain, three samples from North (Chiang Mai), Northeast (Khon Kaen), and Central (Nakhon Pathom) presented one to three mutated genes. LSDV/Thailand/Chiangmai/2021 had a transition mutation from C to T without amino acid change in open reading frame (ORF)133 encoding DNA ligase-like protein, as shown in Fig. 2A. LSDV/Thailand/KhonKaen/2022 had three mutations, including 1) ORF094 encoding putative virion core protein that had a transition mutation from G to A causing amino acid change from Serine (S) to Phenylalanine (F) (Fig. 2B), 2) ORF144 encoding Kelch-like protein that had T deletion, causing an amino acid change from Phenylalanine (F) to Leucine (L) (Fig. 2C), and 3) ORF148 encoding Ankyrin-like protein that had an A insertion in stop codon causing no amino acid change and no ORF change (Fig. 2D). LSDV/Thailand/NakhonPathom/2022 had two mutations, including 1) ORF032 encoding poly(A) polymerase large subunit protein that had a transition mutation from G to A causing no amino acid change (Fig. 2E), and 2) ORF133 encoding DNA ligase-like protein that had a transition mutation from G to A causing an amino acid change from Aspartic acid (D) to Asparagine (N) (Fig. 2F). In contrast, the samples from the West and South did not show any mutations. Overall, the maximum number of mutation genes was three from 156 genes, or less than 2% of the whole genome, as given in Table 1. All the mutation points showed a depth of coverage of at least 10.
DISCUSSION
It is known that LAVs can cause recombination between vaccine viruses and wildtype viruses, resulting in more virulent strains [15] such as canine parvovirus [16], infectious bursal disease virus [17], and bovine herpesvirus type-1 [18]. During outbreaks in South Africa in the 1990s, LSDVs were virulent and vaccine-associated, which showed 67 SNPs compared to attenuated vaccine strains, indicating selection-driven genetic drift after 20 years of LAV implementation [7]. From the current study, it was found that the LSDVs had one to three SNPs after 1.9 years of LAV utilization. A previous study by the authors showed that the number of LSD outbreaks decreased significantly after vaccination [8], which could indicate that the LAVs were still suitable for the control of the disease in Thailand. The LSDV strains from five regions after vaccination were recombinant vaccine strains similar to the Yasothon isolate. The number of mutated genes was less than 2% of the whole genome, which was not surprising because large DNA virus has a low mutation rate [19]. Notably, the duration of the study period, less than two years after vaccination, was relatively short.
While viruses have evolved continuously, the changes in genes coding for replication, structure, and immunomodulator may be shaped upon interaction with the host. ORF032 encoding Poly (A) polymerase large subunit is important for RNA transcription and modification. ORF133 encoding DNA ligase-like protein was mutated in two LSDV strains from Chiang Mai and Nakhon Pathom provinces. This protein is important for DNA replication and nucleotide metabolism. This information is interesting because a recent study utilized this gene to differentiate infection and vaccination (DIVA) [20]. With these mutations, the performance of future assays might change. ORF144 encoding Kelch-like protein is important for virulence and host range, perhaps through the modulation of inflammatory responses [21,22]. Thus, one amino acid change and three amino acids being deleted in this protein could affect the virulence of the Khon Kaen strain. Furthermore, an A insertion in ORF148 encoding Ankyrin repeat protein might change the host range and virulence factors [23]. An amino acid change in ORF094 encoding putative virion core protein might alter the viral structure and infectivity. Based on these findings, morbidity, and mortality rates as well as the prevalence of the disease in these three provinces should be investigated further.
In case of low prevalence, the authors suggested monitoring the gene-encoding proteins for virulence and host range such as Ankyrin repeat protein, Kelch-like protein, and G protein-coupled CC chemokine receptor (GPCR) to optimize the cost and time. A previous study found that a host range gene encoding the Ankyrin repeat protein of five LSDVs in Central and Western Thailand was 100% identical to the Vietnamese strain [9]. However, there were several cases of LSDVs in wildlife such as red bulls, serows, and gaurs in Thailand [9], and camels in India [24]. Therefore, the genes encoding Ankyrin repeat protein for host range determination such as ORF012, ORF145, ORF147, ORF148, and ORF152 should be monitored frequently. This information will be useful for diagnosis in terms of selecting suitable target genes. According to the current study, ORF074 (p32) recommended by the World Organization for Animal Health (WOAH) [13,25] is still appropriate for detecting LSDV due to its conservation, while ORF032 (poly (A) polymerase large subunit), ORF094 (virion core), ORF133 (DNA ligase-like protein), ORF144 (Kelch-like protein), and ORF148 (Ankyrin-like protein) are recommended for mutation analysis.
The virus from the Northeast had the most mutation genes, which might be due to this region having the highest cattle population of around 300,278 heads. This region also suffered from the disease for a longer period than other regions. In contrast, the viruses from the West and South did not show any mutation that might have resulted from having lower animal density and disease prevalence [26]. Indeed, the sample of LSDV from Prachuap Khiri Khan (West) collected during the first LSDV outbreak in 2021 was also researched previously [27]. The virus was almost identical to the Chinese, Vietnamese, and Yasothon strains, indicating low mutation. Another interesting point was that the LSDV strains in Thailand from 2021 to 2022 were different from the strains in Myanmar [28], even though cattle movement across the Thai and Myanmar border continued to occur regularly [29]. This might have been due to the vaccination campaigns and other control measures working effectively.
To the best of the authors’ knowledge, this is the first study of LSDV WGS analysis after vaccination in Thailand. LSDV strains after vaccination with LAVs are recombinant vaccine strains in the same group as the virus that caused the first outbreak in Northeast Thailand. However, the virus from cattle in the North, Northeast, and Central regions had genetic mutations. Thus, annual monitoring of virus genetics is necessary, especially for virulent and host range genes.