
INTRODUCTION
Brain-derived neurotrophic factor (BDNF) is an essential member of the neurotrophin family, that plays a vital role in the development and maintenance of the nervous system [1]. BDNF is expressed across various regions of the male reproductive system, including the testes [2]. Previous studies suggest that BDNF contributes to the maturation and development of germ cells [3,4]. Notably, the identification of BDNF in the Sertoli and Leydig cells and its receptor neurotrophic tyrosine receptor kinase-2 (NTRK2) in spermatogonia of the human [5], bovine spermatozoa [6], ovarian follicles of domestic hen [7] strongly indicates the role of BDNF in the spermatogenesis and gametogenesis. BDNF has been shown to have a significant impact on spermatogenesis. It promotes the proliferation and survival of spermatogonia, as well as the differentiation of spermatocytes into spermatids [8]. BDNF has also been demonstrated to play a role in controlling testicular functions and fertility. The deficiencies in BDNF have been associated with impaired spermatogenesis and decreased fertility [9], conversely, the administration of BDNF has been found to improve spermatogenic functions and fertility in humans [10].
BDNF functions through its high-affinity receptor, NTRK2 (also referred to as TrkB). After binding BDNF to NTRK2, it enhances the NTRK2’s capacity for self-phosphorylation and activates the downstream signaling pathways, causing a change in the gene expression and modulation of germ cell activities [11]. After NTRK2’s self-phosphorylation, the RAS-mitogen-activated protein kinase pathway gets activated, which, in turn, promotes cell division and proliferation, while the phosphoinositide-specific phospholipase C pathway induces inositol triphosphate activation, which further increases the intracellular calcium release and ultimately improves the synaptic plasticity of neurons [12]. These interactions between BDNF and NTRK2 are known to promote several critical physiological processes involved in reproduction, including cellular adhesion, angiogenesis, resistance to apoptosis, and cellular proliferation [13–17].
In the context of stallions, it is important to note that these animals exhibit a distinct pattern of seasonal breeding. This pattern involves a decrease in sperm production throughout the non-breeding season (NBS) [18]. During the NBS, spermatozoa production along with gonadotropin and testosterone concentrations decreased [19]. This decline has a significant impact on the horse breeding industry affecting both breeding strategies and the financial objectives. Therefore, contemporary management of stallion reproduction emphasizes collecting semen during times that fall beyond the conventional breeding season (BS) [20]. There is a dire need for time to study and identify the potential internal and external factors that may contribute to maintaining the optimum spermatogenesis throughout the year and the fertility of stallions.
As a consequence of the implications that BDNF and NTRK2 have in the reproductive processes, there is a pressing need to investigate the expression of BDNF and NTRK2 in the seasonal breeding pattern of stallions. We hypothesized that both BDNF and NTRK2 are present in testicular cells of stallion and their expression pattern varies depending upon the season of the year. Therefore, this study was conducted to investigate the localization and seasonal variation in the expression pattern of BDNF and its receptor NTRK2 in stallions’ testes.
MATERIALS AND METHODS
A total of eight stallions (Thoroughbred) were selected and assigned to two groups—BS (n = 4; age 36 ± 4.0 months; castration months: June-July) and NBS (n = 4; age 36 months; castration month: January) according to the season of the year. The castration of these animals was performed in the field after getting consent and as per the desire of the farmers by a registered veterinary surgeon. The researchers were not present at the castration site. Therefore, the Kyungpook National University’s Animal Experiment Ethics Committee or any other comparable animal ethics body did not need to approve this work.
The preparation and preservation of testicular tissues of the stallions were performed as previously described [21] with some modifications. In brief, the testes obtained after the castration were placed immediately in an ice box maintained at 4°C and then shifted to the laboratory. During the dissection and histological observation of the testicular tissues, a careful examination was performed. For the tissue fixation analysis and reverse real-time quantitative polymerase chain reaction (RT-qPCR), the testes were sliced into multiple sections. For the immunofluorescence microscopy, the tissue (approximately 1.0 cm3) was immersed in 4% paraformaldehyde at room temperature for 24 h. After thorough washing with phosphate-buffered saline (PBS) for 24 h, the tissues were dehydrated in a successive ethanol dilution series before embedding in a paraffin block. For RT-qPCR, approximately 0.5 cm3 of testicular tissues were instantly snap-frozen for 1 min in liquid nitrogen at −196°C and stored at −80°C till further analyses.
The RNA extraction from the testicular tissues of Thoroughbred stallions was performed. Briefly, 300 µL of the TRI reagent solution (Cat. #01066257, Thermo Fisher Scientific, Waltham, MA, USA) was mixed with 1 g of testicular tissues and chopped using a microtube electric mixture (1-8505-01, Labotech, Daejeon, Korea). After homogenizing the sample, 700 µL of the TRI reagent solution was added, followed by 200 µL of chloroform (C2432, Sigma Aldrich, St. Louis, MO, USA). Following vertexing and incubation for 5 min at room temperature, the samples were centrifuged for 15 min at 2,000×g and 4°C. The supernatant containing the RNA was collected and mixed with an equal amount of iso-propanol (K4T116, Duksan Pure Chemicals, Ansan, Korea). The samples were stored for 3 h at −20°C and centrifuged for 20 min at 2,000×g and 4°C. The supernatant was discarded, and the pellet was rinsed with 700 µL of 70% ethanol (FAEL61, Duksan Pure Chemicals) and re-centrifuged for 10 min at 2000 g and 4°C. Finally, after drying the sample, the pellet was mixed with 50 µL of RNase-free water (10977-015, Life Technologies, Carlsbad, CA, USA). The concentration of RNA was measured by NanoDrop (Bio Tek Instruments, Winooski, VT, USA), and the isolated RNA was stored at −80°C for further processing.
The PrimeScriptTM 1st strand cDNA Synthesis kit (cat. no. 6110, Takara Biotechnology, Shiga, Japan) and oligo-dT primers were utilized for the cDNA synthesis. The first solution consisted of 8 µL of the RNA solution combined with RNase-free water, 1 µL of oligo-dT primer, and 1 µL of dNTP. The solution was added to the SimpliAmp Thermos Cycler (Thermo Fisher Scientific) following centrifugation at 1,000×g for 1 min. At 4°C, 10 µL of the second solution was added, which included a combination of 4 µL of 5× prime script buffer, 0.5 µL of the RNase inhibitor, 1 µL of the prime script RTase, and 4.5 µL of RNase-free water. Finally, 140 µL of ultrapure water was added to dilute the cDNA solution and stored at −20°C until further processing.
With some modest adjustments from the previously described procedure [22], RT-qPCR analysis of BDNF and NTRK2 was performed. The qPCR reaction mixture consisted of a total volume of 20 µL, which included 8 µL of cDNA solution, 10 µL of Power SYBR® Green PCR Master Mix (cat. no. 4367659, Applied Biosystems; Thermo Fisher Scientific), 1.6 µL of RNA-free ultrapure water, and 0.2 µL of reverse and forward primers each. A list of the reverse and forward primers employed in the investigation is provided in Table 1. The RT-qPCR reaction was performed with the StepOnePlus™ Real-Time PCR System (Applied Biosystems) under the following amplification conditions: initial denaturation at 95°C for 10 min, followed by 40 cycles of denaturation at 95°C for 15 s and annealing at 60°C for 1 min. The melt curve stage included denaturation at 95°C for 15 s, annealing at 60°C for 1 min, and denaturation at 95°C for 15 s. The 2−ΔΔCt protocol was employed for data analysis [23], the results presented as fold-changes, and the mRNA levels of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were utilized to normalize the relative mRNA transcript abundance of BDNF and NTRK2.
Western blotting (WB) was performed to validate the cross-reactivity of BDNF and NTRK2 rabbit polyclonal antibodies. All the primary and secondary antibodies with their dilution rates used for WB and immunoflourence assay are listed in Table 2. Previously reported protocol [24] of WB was adopted, albeit with minor modifications. Briefly, at the time of sample collection, the tissues of testicular parenchyma were stored at −80°C after snap-freezing in liquid nitrogen. After thawing the testicular tissues, the sample was placed into radioimmunoprecipitation assay (RIPA) buffer solution along with protease inhibitor (Roche, Penzberg, Germany) and homogenized with the Polytron PT 1200 CL homogenizer (DAIHAN Scientific, Wonju, Korea). Each sample’s protein concentration was evaluated by using an absorbance microplate reader (Tecan, Männedorf, Switzerland) with a filter of 560 nm wavelength. The homogenized testicular tissue samples were diluted at a concentration of 2 mg/mL with the RIPA buffer solution. For 15 min, the protein samples were heated in boiling water and diluted (1:1 ratio) with the sample buffer solution (Laemmli sample buffer, Bio-Rad, Hercules, CA, USA). The diluted samples were loaded (15 µL) into a 10% sodium dodecyl sulfate (SDS)-poly acrylamide gel and separated by using the Mini-Protean II Electrophoresis System (Bio-Rad). The protein was then transferred by the Mini Protean Tetra System (Bio-Rad) onto the AmershamTM ProtranTM 0.2-µm nitrocellulose blotting membrane (GE Healthcare, Düsseldorf, Germany). Blotting was performed by using blotto milk (PBST having 5% DifcoTM skim milk, Difco, Saint-Ferreol, France) at room temperature for 45 min. The membranes were then incubated with BDNF and NTRK2 antibodies diluted in blotto reagent overnight at 4°C. Normal rabbit IgG was used with the same dilution rate as primary antibodies to serve as a negative control and for control positive, mouse monoclonal β-actin antibody was used. For secondary antibodies, horseradish peroxidase-conjugated anti-rabbit IgG diluted in blotto milk was used and incubated for 1 h at room temperature. The blots were washed thrice with 1X PBST buffer. Enhanced chemiluminescence detection reagents (catalog no. 34580, Thermo Fisher Scientific) were used to identify, and ImageQuant LAS 500 (Cytiva) was used to partially quantify the protein bands. The ImageJ software was used to examine the relative intensity of BDNF and NTRK2 protein bands, which were normalized to β-actin.
The immunofluorescence labeling of BDNF and NTRK2 was performed on the stallion testicular tissues as described earlier with minor alterations [21]. Briefly, the slides with testicular tissue slices of around 5 µm were affixed and kept at 4°C. After xylene deparaffinization (Duksan Pure Chemicals), the slides were rehydrated in a succession of ethanol concentrations (100%, 95%, 80%, 70%, 50%, and 25%). Antigen Retrieval Buffer (100X citrate buffer, pH 6.0, Abcam, Cambridge, UK) was applied to the tissue slides for 30 min at 97.5°C. The tissue slides were then washed twice in PBST for 2 min each time and blocked with a PBS solution containing 5% donkey serum (Sigma Aldrich) for 30 min after cooling to room temperature. In a blocking solution of PBS containing 5% donkey serum, the BDNF and NTRK2 rabbit polyclonal antibodies were employed. For the negative control, normal rabbit IgG was used at the same dilution as the primary antibodies. These were incubated with the tissue sections for 1.5 h in a humid chamber and then washed with PBST for 5 min (×3). The tissue sections were then incubated with secondary antibodies at room temperature in a humid chamber for 45 min, followed by washing with PBST for 5 min (×3). After mounting with the Vectashield® mounting media containing 4,6-diamidino-2-phenylindole (Vector Laboratories, Burlingame, CA, USA), coverslips were placed over the tissue slices. The preparation was finally sealed with transparent viscous fluid (Estée Wannabe, Seoul, Korea).
Using the DM2500 fluorescent microscope (Leica, Wetzlar, Germany), cells immunolabeled with BDNF and NTRK2 were observed. An external light source, EL 6000, was installed in the microscope (Leica). A dual-emission filter for tetramethyl rhodamine isothiocyanate and fluorescein-5-isothiocyanate was employed to investigate the composite fluorescence expressions. For BDNF and NTRK2, cells with green fluorescence were considered positive, whereas those without any fluorescence were considered negative. The photographs that were immunolabeled were taken with the Leica DFC 450 C Digital Camera.
All data were analyzed using SPSS Statistics for Windows, version 25 (IBM, Armonk, NY, USA). Levene’s test for equality of variance and Shapiro-Wilk test for normal distribution of data was performed. The non-parametric test (Mann-Whitney test) was used to find the significance level of mRNA transcripts of BDNF and NTRK2 between different seasons. The seasonal difference between the relative intensity of protein bands of BDNF and NTRK2 was evaluated using the independent sample t-test. p ≤ 0.05 was regarded as significant, and the data represents the standard error of the mean (± SEM).
RESULTS
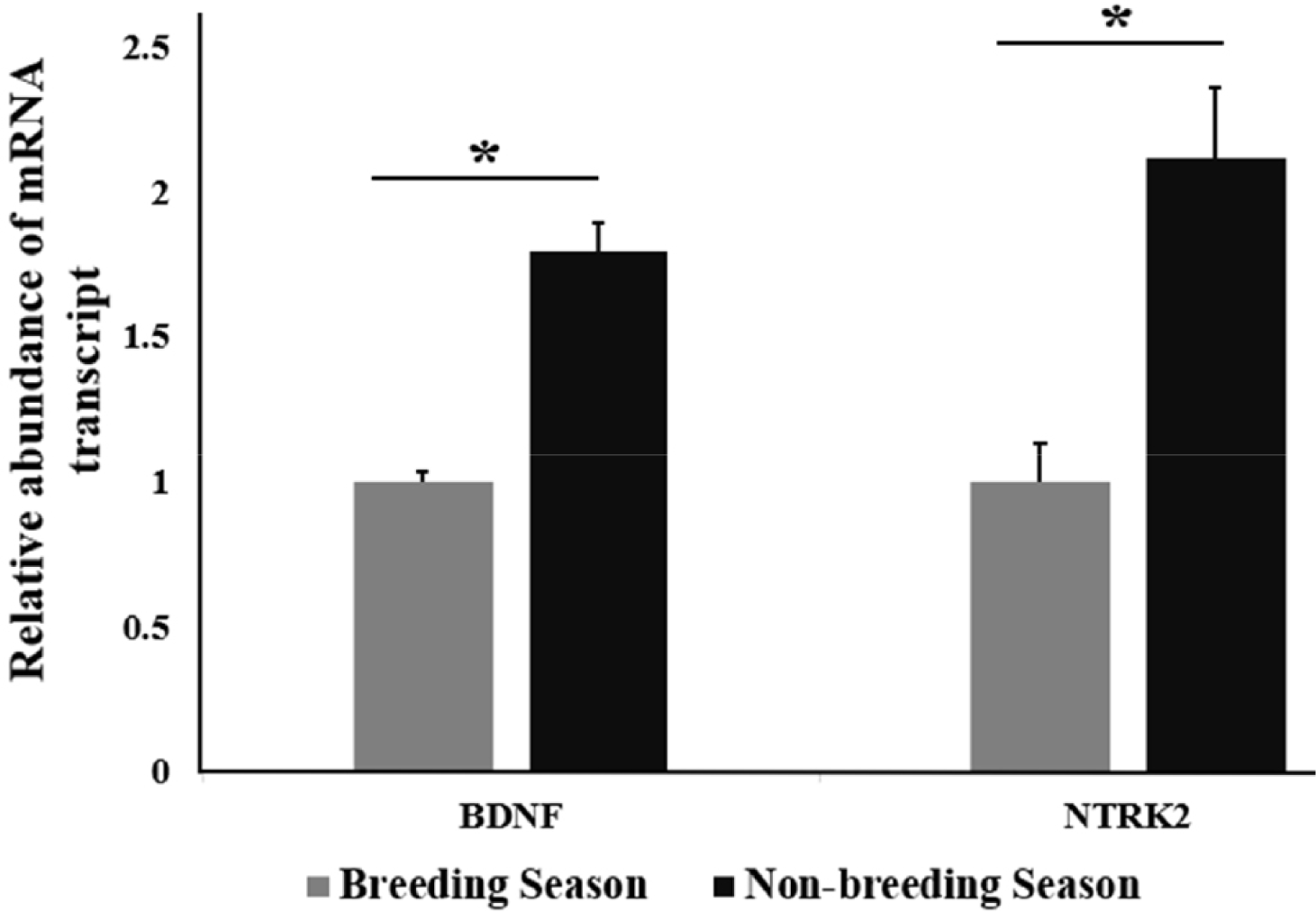
By using RT-qPCR analysis, the relative abundance of mRNA transcript of BDNF and NTRK2 in stallion testes from different seasons, including the BS and NBS was determined. A significant upregulation of the relative abundance of BDNF mRNA transcript was found in NBS (p < 0.05) compared with the BS. A significant (p < 0.05) upregulation of the relative abundance of mRNA transcript of NTRK2 was recorded in the NBS (Fig. 1) compared to that in the BS.
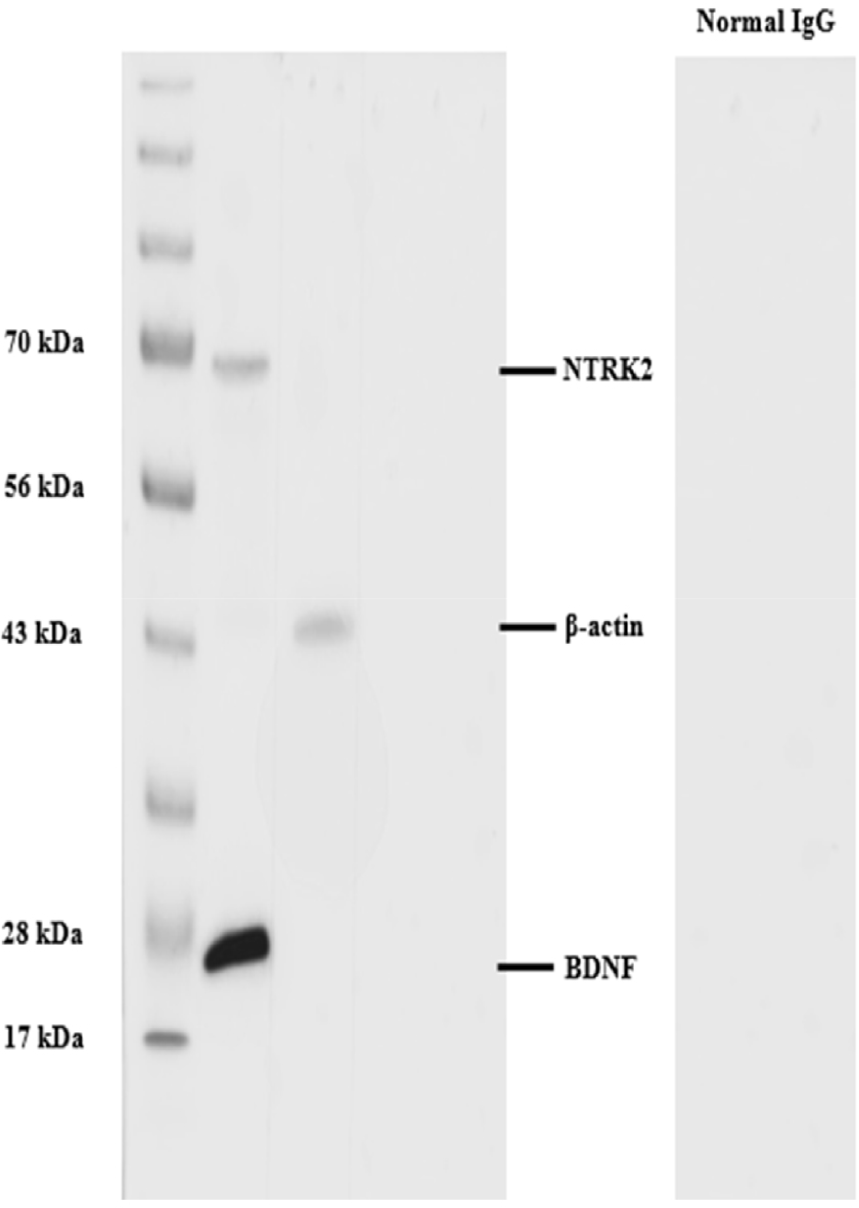
To examine the cross-reactivity of the stallion testicular tissues with BDNF and NTRK2 antibodies, WB was performed. The BDNF and NTRK2 proteins were identified to have molecular weights of around 26 kDa and 68 kDa, respectively (Fig. 2). The protein band of control positive β-actin was observed at 43 kDa. The negative controls that received rabbit IgG instead of the primary antibody did not exhibit the band (Fig. 2).
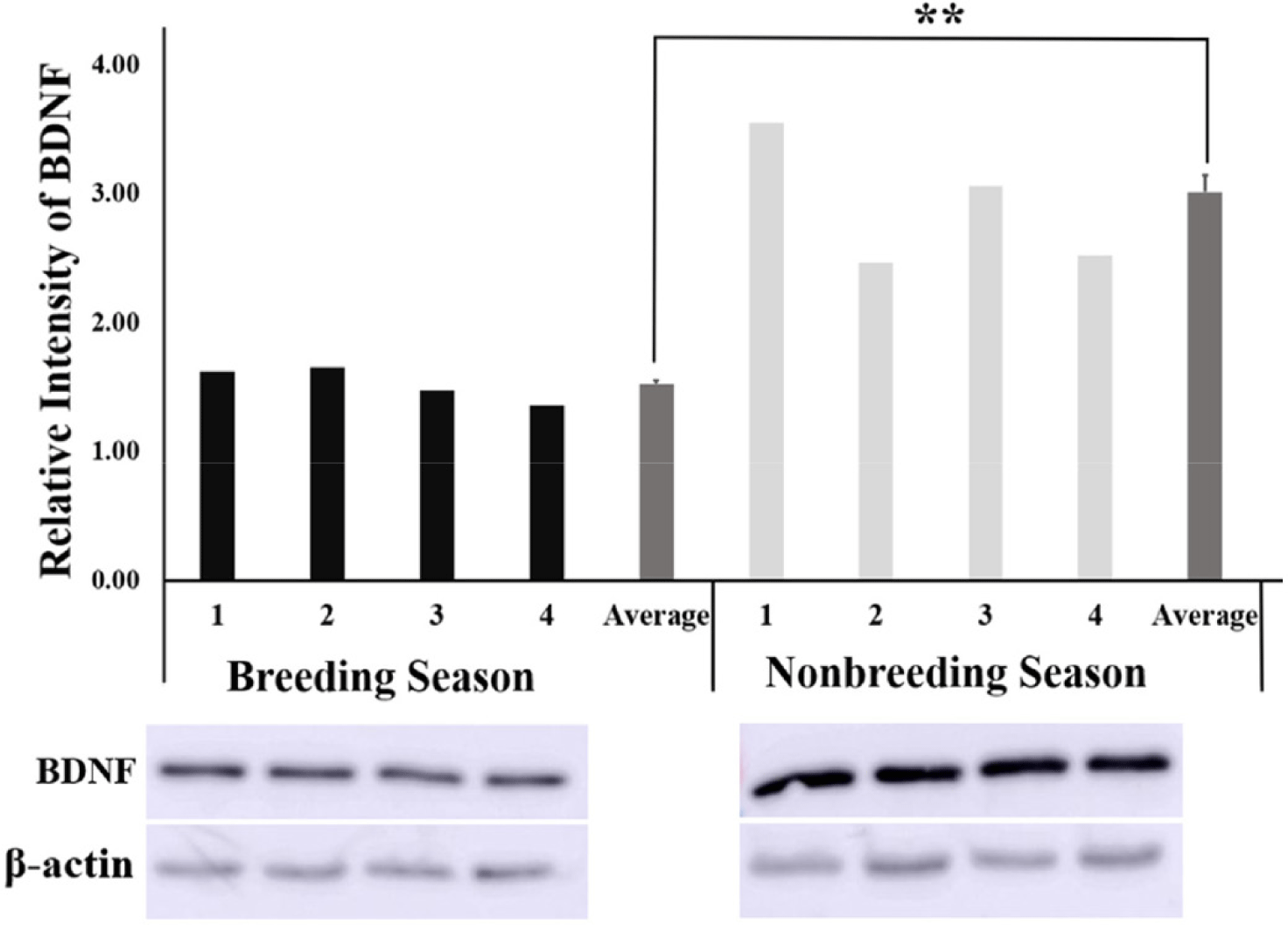
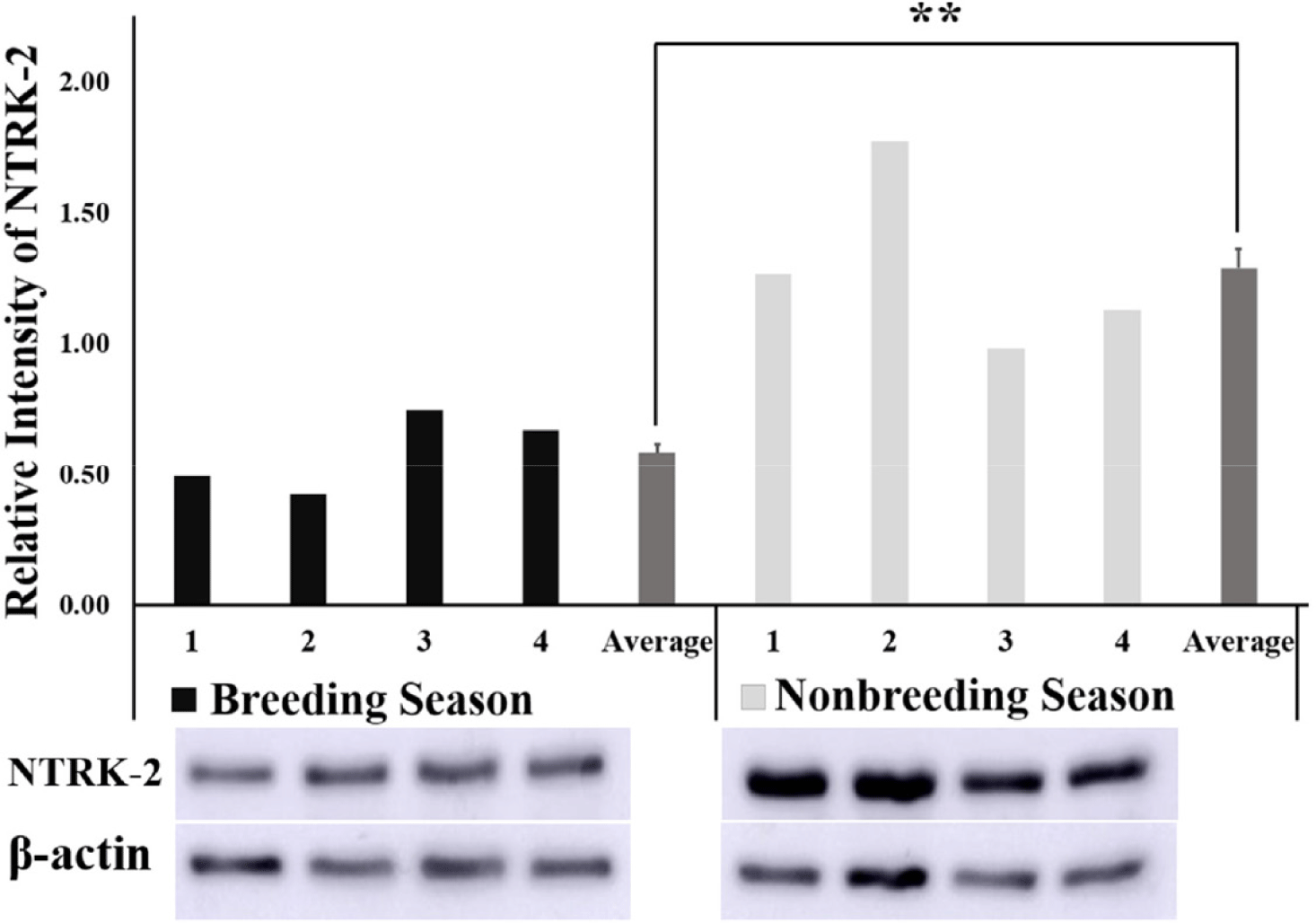
The relative intensity of BDNF obtained from the quantification of protein bands using the ImageJ software was found to be significantly higher (p < 0.01) in the NBS than in BS (Fig. 3) Similarly, the relative intensity of the NTRK2 protein bands was significantly higher (p < 0.01) in the NBS compared with that of BS (Fig. 4).
The immunolocalization of BDNF-positive cells in the stallion testes from BS and NBS was investigated. The cytoplasm of Sertoli and Leydig cells were stained with BDNF antibody (Fig. 5). Sertoli cells were immunolabeled with BDNF in the BS (Figs. 5A and 5D). Germ cells were not immunolabeled with BDNF in the BS. It was also observed that the cytoplasm of a few spermatogonia was stained in the NBS (Figs. 5F and 5H). The normal rabbit IgG stained with the same concentration as BDNF displayed no immunolocalization in any type of testicular cells in both groups (Figs. 5E and 5J).
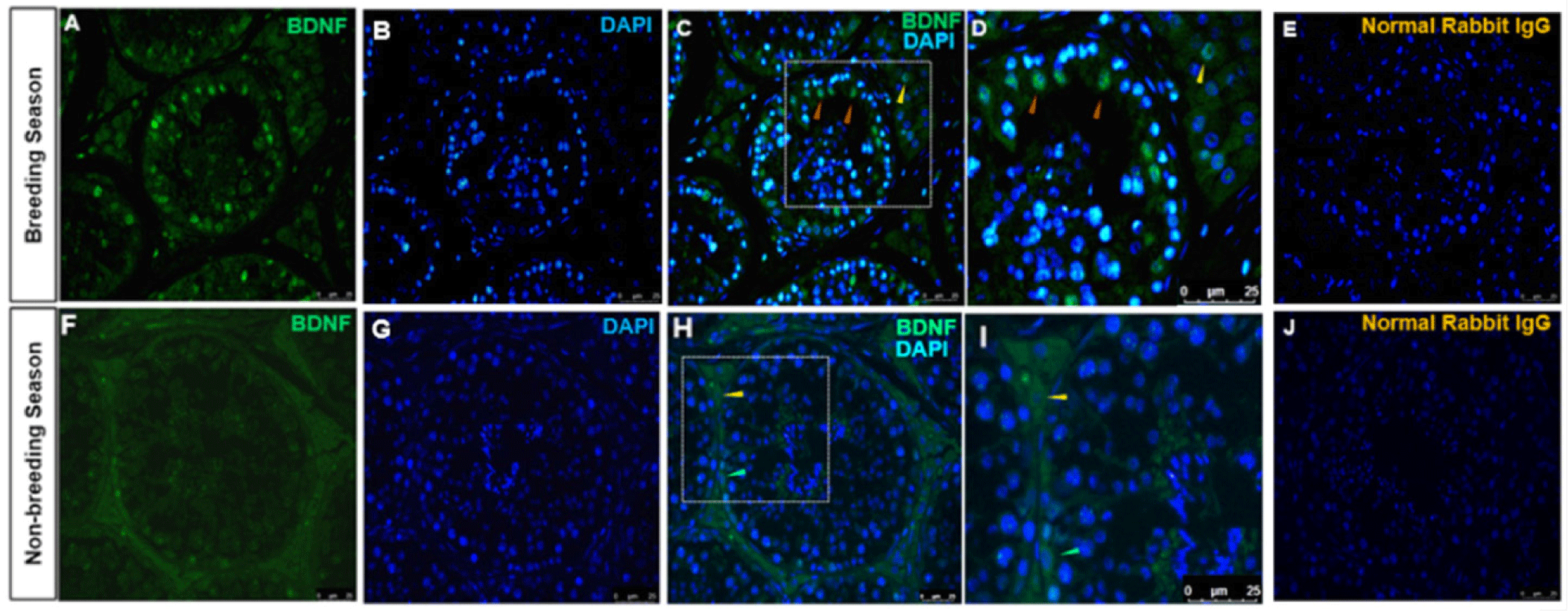
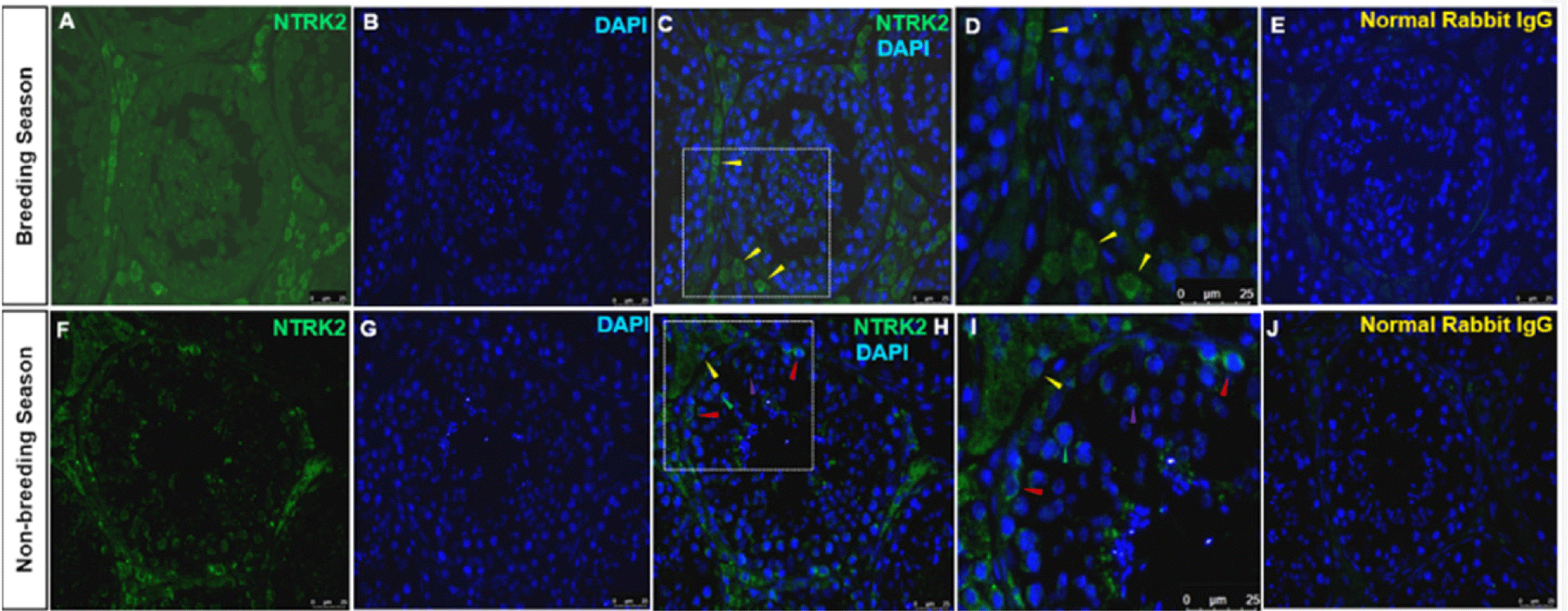
Immunolocalization of the NTRK2-positive cells was observed mainly in Leydig cells during the BS and the NBS. No Sertoli or germ cell staining was detected in the BS. In the NBS, the localization was identified in the Leydig cells’ cytoplasm and different stages of spermatogonia including undifferentiated spermatogonia, primary and secondary spermatocytes (Fig. 6F). No Sertoli cells were stained in the NBS. The normal rabbit IgG stained with the same dilution as NTRK2 displayed no immunolocalization in any type of the testes cells (Figs. 6E and 6J).
DISCUSSION
BDNF and its receptor NTRK2 played a vital role in the differentiation, proliferation, and survival of neural cells [25]. For improving knowledge of reproductive biology, particularly spermatogenesis, and the possible non-neuronal activities of these neurotrophins, it is crucial to study the localization and seasonal expression variations of BDNF and NTRK2. Our present results revealed the expression of BDNF and NTRK2 in the Sertoli, Leydig, and germ cells of the stallion testes. We also reasonably investigated that BDNF and its receptor NTRK2 were seasons- and location-dependent in the stallion testes.
In our study, we identified the localization of BDNF in the cytoplasm of Sertoli and Leydig cells and the early stages of spermatogonia. These results are in line with a previous study where Chan Park and coworkers investigated the expression of different neurotrophins in mice testes. They found that NT-3 was expressed in the spermatocytes and spermatogonia while the BDNF was expressed in the Sertoli cells [3]. Hence, the secretion of BDNF by Sertoli cells potentially serves as a trophic factor, promoting the survival of spermatogonia throughout their differentiation phase. Another study revealed the expression of BDNF during testes morphogenesis in the prenatal and adult human testes. They found that BDNF was expressed in Leydig cells predominantly with lower intensity of expression in Sertoli cells and spermatocytes [5]. Researchers from Italy investigated the expression of BDNF in type B spermatogonia in zebrafish [26]. Moreover, localization of BDNF in the head, neck, and tail of spermatozoa has been studied in bovine [6] and humans [27]. In addition to controlling apoptosis and sperm viability, BDNF is essential for mitochondrial function. In ejaculated sperm, it increases insulin and leptin release, enhancing cell viability [28]. The presence of BDNF in the spermatogonia, Sertoli, and Leydig cells in stallion testes in our study and previous works suggested that BDNF may be involved in the steroidogenesis, spermatogenesis, maturation, and morphogenesis of the stallion testis however, more research is warranted to identify the precise function of BDNF in these processes.
In this experiment, the localization of NTRK2 was detected in the cytoplasm of Leydig cells, and different stages of spermatogonia, including undifferentiated spermatogonia, primary and secondary spermatocytes. Our results are consistent with previous work, wherein NTRK2 immunoreactivity was detected in somatic cells (Leydig and Sertoli cells), spermatogonia, and spermatids in mice [3,29]. In a study conducted on a mouse tumor Leydig cell line, an increased cellular steroid synthesis caused by nerve growth factor exposure suggests that neurotrophins play a role in differentiating processes [5]. Research led by Safari and colleagues demonstrated that exogenous BDNF had a notable impact on the viability, motility, nitric oxide concentration, mitochondrial activity, and lipid peroxidation content of human spermatozoa [10]. Insulin and leptin secretion by bovine sperm were increased when cells were exposed to exogenous BDNF, whereas insulin was decreased by K252a (NTRK2 inhibitor), inferred that BDNF could be a regulator of sperm secretion of insulin and leptin through the NTRk2 receptor as the sperm viability and mitochondrial activity were both decreased when the BDNF/NTRK2 signaling pathway was blocked with K252a [6]. The testosterone synthesis and Leydig cell activity may be controlled by the BDNF/NTRK2 signaling pathway. Moreover, the activation of NTRK2 increases the expression of the steroidogenic acute regulatory protein (StAR), a crucial enzyme involved in testosterone production in Leydig cells [30]. The presence of BDNF/NTRK2 in somatic and germ cells led to the hypothesis that the Sertoli cells release BDNF, which interacts with spermatogonial stem cells and regulate spermatogenesis. Furthermore, the immunolocalization of NTRK2 in Leydig cells indicates the potential role during testes morphogenesis and steroidogenesis.
Our study also examined how the expression of BDNF and NTRK2 varies seasonally. Both the intensity of protein bands and mRNA transcript abundance of BDNF and NTRK2 were higher in the NBS compared to the BS. We found that BDNF was present in the cytoplasm of spermatogonia specifically during the NBS and NTRK2 was expressed in various stages of spermatogonia, including undifferentiated spermatogonia, primary spermatocytes, and secondary spermatocytes, during the NBS. There are two possible interpretations for these findings: one theory suggests that during the BS, when there are adequate levels of follicle-stimulating hormone (FSH) and luteinizing hormone (LH), spermatogenesis proceeds at a normal pace, requiring less BDNF. However, during the NBS when FSH and LH levels are lower [31]; more BDNF is needed to maintain the minimal level of spermatogenesis, leading to additional BDNF expression in germ cells during this period. Another interpretation for this higher expression during the NBS could be that spermatogenesis reaches its peak during the BS, and testicular cells like Leydig, Sertoli, and germ cells use BDNF and NTRK2 extensively to support efficient spermatogenesis. However, during the NBS, when spermatogenesis decreases by 50% in stallions [32], the testes retain BDNF and NTRK2 as reserves. The data about the seasonal variation of the expression pattern of NTRK2 is scanty, however, during male germ cell development [5], BDNF and NTRK2 expression are variable and depend upon specific times and locations. Based on these findings, BDNF seems to be a promising neurotrophin that may contribute to stallion fertility. Further research such as investigating the functional impact of BDNF signaling on spermatogenesis and examining the regulatory mechanisms controlling the observed seasonal expression patterns is warranted.
CONCLUSION
In conclusion, the BDNF and its high-affinity receptor NTRK2 are responsible for a wide variety of crucial activities in male reproduction. We reasonably investigated that BDNF and its receptor NTRK2 immunolabeling were seasons- and location-dependent in stallions’ testes. A deeper comprehension study of the molecular mechanism regulating the BDNF/NTRK2 signaling may provide new insights and help the horse breeding industry by maintaining stallion fertility throughout the year.