
INTRODUCTION
Porcine epidemic diarrhea (PED) is a highly contagious enteric disease characterized by acute symptoms such as watery diarrhea associated with vomiting and dehydration [1,2]. The most severe signs have been reported in piglets less than two weeks old, in which diarrhea leads to severe dehydration and is associated with mortality rates of up to 100% in affected litters [3,4]. PED is mainly caused by the porcine epidemic diarrhea virus (PEDV), a member of the family Coronaviridae, and is characterized by a positive-sense, enveloped single-stranded RNA virus [1,4]. PEDV contains a 28 kb positive-sense single-stranded RNA genome with a 5′ cap and 3′ polyadenylated tail. The PEDV genome encodes structural and non-structural viral proteins, such as spike (S), membrane (M), and nucleocapsid (N) proteins, which are important for viral infection, replication, and immune response evasion [2,5]. Owing to their ability to mount a sufficient immune response, these proteins are crucial for the development of effective vaccines [5]. Live attenuated and inactivated vaccines are the most common immunization methods for PEDV. In contrast, the use of conserved epitopes of pathogen proteins in subunit vaccine design is gaining interest because of its immunogenicity, safety, and cost-effectiveness compared to traditional vaccines [6].
The PEDV M protein, a prevalent component of the viral envelope, is a triple-spanning structural membrane glycoprotein featuring an exterior short amino-terminal domain and an interior long carboxy-terminal domain [2,7]. This protein interacts with the S and N proteins and plays an important role in the assembly of viral particles [8,9]. In addition, antibodies targeting the M protein of coronaviruses are crucial for controlling the course of the disease and inducing protection against the virus [10,11]. Meanwhile B-cell epitopes have been widely used in the development of antibody-based therapies, peptide-based vaccines, and immunodiagnostic tools [9,12]. Progress in B-cell epitope mapping and computational prediction using bioinformatics tools have provided molecular understandings of bio-recognition process and antigen-antibody complex formation, leading to the development of more accurate algorithms for predicting antigen localization. [13]. Identification of epitopes on the PEDV M protein is also valuable for elucidating its antigenic properties [9].
Lactic acid bacteria (LAB) have attracted attention not only because they are safe to use but also for their capability to colonize the intestines, withstand gastric and bile acids, and produce anti-microbial substances [14–19]. Moreover, LAB are considered attractive candidates for mucosal vaccine delivery vehicles owing to their intrinsic adjuvanticity, long history of use in dairy and other fermented foods, and their inclusion in the Generally Recognized as Safe (GRAS) list [20]. The cell surface display of heterologous proteins on LAB is a growing research field that shows great potential for a variety of applications, including the development of live vaccine delivery system, screening peptide libraries, and developing whole-cell biocatalysts [20–22]. Recent research has shown the promising application of LAB as mucosal vaccine delivery vehicles. Hou et al. [23] successfully displayed the PEDV N protein on the surface of Lactobacillus acidophilus. Several studies have demonstrated the ability of the core neutralizing epitopes of the PEDV surface displayed proteins on Lb. casei and Lb. johnsonii to elicit immune response [24–28]. Zang et al. [29] and Li et al. [30] used the S proteins of PEDV and displayed them in Lb. acidophilus and Lb. casei, respectively.
Although many studies have reported the application of S and N proteins in PEDV, studies exploring the use of M proteins and their immune properties are limited [5]. Moreover, the immunogenicity of surface-displayed PEDV M epitopes in LAB has not been investigated. In this study, we predicted the B-cell epitopes for the PEDV M protein and developed a surface display platform utilizing the epitopes of the PEDV M protein in Lactiplantibacillus plantarum SK156. Innate responses in porcine intestinal epithelial cells (IPECs) and immune responses elicited following oral vaccine administration in mice have also been described.
MATERIALS AND METHOD
Membrane protein sequences of PEDV isolated in Korea (KOR/KNU-141112/2014; GenBank accession no. ADZ76336), Japan (OKY-1/JPN/2014; GenBank accession no. LC063847), China (CH/JSX/06; GenBank accession no. EU033967), and Belgium (CV777; GenBank accession no. AF353511) were accessed from the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/), and conserved regions were compared using ClustalW on MEGA6 (http://www.megasoftware.net/). Prediction of protein structure was performed using trRosetta web-based tool [31] and evaluated using ProSA-web [32]. Prediction of linear B-cell epitopes were performed using three tools: IEDB Bepipred Linear Epitope Prediction 2.0 tool [33] and SVMTriP [34]. These three tools employ different models, such as the Hidden Markov and Support Vector Machine models, and consider different amino acid propensities and secondary structures to predict B-cell epitopes [35]. In the IEDB tool, other epitope properties such as surface accessibility (Emini surface accessibility) and antigenic propensity (Kolaskar and Tongaonkar antigenicity), were also used to select the epitopes. The results from each tool were compared and the most conserved immunogenic epitopes were chosen for surface display on Lp. plantarum.
Table 1 provides a summary of the bacterial strains and plasmids utilized in this study. Escherichia coli DH5α was propagated in the Luria-Bertani (LB) broth (Difco, Davenport, IA, USA) supplemented with ampicillin (100 μg/mL) when applicable under shaking conditions at 37°C. The lactobacilli strains were cultivated in the de Mann, Rogosa, and Sharpe (MRS) broth (Difco) and grown without agitation at 37°C. Erythromycin (3 μg/mL) was added when applicable.
| Strain or plasmid | Features or sequence | Source or reference |
|---|---|---|
| Strains | ||
| Lactiplantibacillus plantarum SK156 | Host for transformation, erythromycin resistance- negative | [72] |
| Lactobacillus acidophilus ATCC 4356 | Source of surface layer protein A | [36] |
| Escherichia coli DH5α | Host for transformation | TaKaRa Bio (Japan) |
| Plasmids | ||
| pULP3:SP:GFP:CWA | pULP2:PLDH with SP+GFP+CWA fusion gene | This study |
| pULP3:SP:M1:CWA | pULP2:PLDH with SP+PEDV M protein epitope 1+CWA fusion gene | This study |
| pULP3:SP:M2:CWA | pULP2:PLDH with SP+PEDV M protein epitope 2+CWA fusion gene | This study |
| pULP3:SP:M3:CWA | pULP2:PLDH1 with SP+PEDV M protein epitope 3+CWA fusion gene | This study |
Primers listed in Table 2 were used to amplify the DNA sequences encoding the signal peptide (SP) and cell wall anchor (CWA) domain of surface layer protein A (SlpA) from Lb. acidophilus 4356 [36]. Likewise, PEDV epitopes designated as M1, M2, and M3 were amplified from the PEDV strain KVCC-VR0000187 using the primers listed in Table 2. Purified polymerase chain reaction (PCR) products (SP, CWA, and M epitopes) were used to perform recombinant PCR using the primers listed in Table 2. The DNA fragments obtained were designated as SP-M1 epitope-CWA (M1), SP-M2 epitope-CWA (M2), and SP-M3 epitope-CWA (M3) fusion genes. The fusion genes and pULP3 were digested with PstI and HindIII, respectively, and ligated with T4 DNA ligase (TaKaRa Bio) for bacterial transformation. E. coli DH5α transformation was done following previous protocol [21]. Lactobacillus transformation was performed using electroporation as described by Chae et al. [37]. Transformants were selectively grown using the appropriate media: LB agar with ampicillin (100 µg/mL) or erythromycin (150 µg/mL) for E. coli, and MRS agar supplemented with erythromycin (3 µg/mL) for Lp. plantarum strain. The transformants were grown at 37°C for 12–18 h (E. coli), or 48–72 h (Lp. plantarum).
The expression of the PEDV M epitope was determined as previous protocol [21]. Briefly, recombinant E. coli BL21 (DE3) cells carrying the M epitope genes (optical density [O.D.600] = 0.6) were overexpressed by adding 0.1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) and incubated at 25°C for 6 h. Then, the cells were centrifuged at 10,000×g for 10 min and resuspended in Tris-Cl buffer (50 mM Tris, pH 8.0). Cells were lysed using a probe-tipped sonicator (Vibra-Cell) set at 30% amplitude 15 times for 10 s each with 10-s interval on ice. The suspension was centrifuged at 13,000×g for 20 min and the pellet was collected and washed twice with lysis buffer. The pellets were solubilized in 8 M urea. Protein purification was performed using Ni-NTA agarose-packed columns (Qiagen). For western blotting, the purified proteins were separated on a 12% polyacrylamide gel. Proteins were subsequently transferred to nitrocellulose membranes. (Bio-Rad). After blotting, the membrane was washed with 1× Tris-buffered saline containing 0.1% Tween 20 (TBST) and blocked with 5% bovine serum albumin (BSA; R&D Systems) in TBST for 1 h at room temperature. Monoclonal anti-His antibody (1:20,000 dilution in TBST with 5% BSA) was added and incubated overnight at 4°C. The membrane was washed with TBST before incubation with horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (1:20,000 dilution in TBST with 5% BSA) (Thermo Scientific) for 1 h at room temperature. Proteins were detected using the SuperSignal West Pico Chemiluminescent Substrate kit (Thermo Scientific) and observed using ChemiDoc XRS+ and Image Lab software (Bio-Rad).
Recombinant Lp. plantarum SK156 was incubated overnight in MRS broth with erythromycin (3 µg/mL). The immunofluorescence assay was performed according to Hwang et al. [38]. Briefly, cells were incubated and grown at 37°C for 12 h and then harvested by centrifugation. Subsequently, the cells were washed with chilled phosphate buffered saline (PBS; pH 7.4) and reconstituted in an equivalent volume of the same buffer. Multi-well glass slides were prepared, and 10 μL poly L-lysine solution (0.1% w/v; Sigma-Aldrich) was added to each well. The mixtures were incubated for 1 h and the liquid was aspirated off. The cells were washed once with sterile distilled water and air dried. The cells were treated and rinsed with PBS containing 0.1% (v/v) Tween-20 (PBST, pH 7.4), then blocked with 2% (w/v) BSA in PBST buffer for 30 min at room temperature. The solution was then aspirated off and 10 μL of diluted (1:200) primary antibody (anti-HisTag antibody; R&D Systems) dissolved in 2% BSA with PBST buffer was added. The slide was incubated overnight at 4°C and washed with PBST. Subsequently, the cells were incubated with secondary antibody in PBST (NorthernLights Anti-mouse IgG-NL557; R&D Systems) with 2% BSA for 1 h at room temperature in the dark. The secondary antibody solution was decanted and washed thrice with PBST for 5 min each in the dark. Finally, the bacterial cells were reconstituted in a mounting solution. The cells were viewed under a fluorescence microscope (ProgRes C10 plus with Intensilight C-HGFI; Nikon) equipped with a 570 nm filter.
IPEC-J2 was grown using Dulbecco’s modified Eagle medium (DMEM; Gibco) in a humidified atmosphere with 5% CO2 at 37°C [39]. The IPEC-J2 cells were seeded in 24-well plates and allowed to reach at least 90% confluence. Cell concentration was determined using 0.4% trypan blue viability staining. The wild-type and recombinant Lp. plantarum SK156 displaying M epitopes on its surface were prepared at approximately 2.5 × 107 CFU/mL and re-suspended in DMEM. Bacterial cells were incubated with IPEC-J2 cells for 2 h. Later, cell culture supernatant was collected and stored at −70°C until assayed.
The Institutional Animal Ethics Committee of Dankook University in Korea approved all animal experimental procedures (DKU-16-038). Thirty (30) female, specific pathogen-free BALB/c mice (8-weeks old) were purchased (Raonbio, Yongin, Korea) and adapted to the laboratory environment for 1 week (Fig. 5A). The animal room had a 12-h light-dark cycle and kept at 22°C–25°C with 45%–50% relative humidity. Mice were given unrestricted access to a standard pellet diet (Envigo) and sterilized distilled water. After acclimatization, the mice were randomly divided into five groups (six mice per group, three mice per cage). Immunization was performed by oral gavage (0.1 mL) containing PBS only (pH 7.4, control), wild-type Lp. plantarum SK156 without M epitopes in PBS (SK156), and 2×109Lp. plantarum SK156 cells expressing PEDV M epitopes (M1, M2, or M3). Oral immunization was performed for three consecutive days, on days 0–2, 14–16 (first booster), and 28–30 (second booster). Blood samples were collected from the tail vein on days 0 (pre-immune), 21, and 35. Serum samples from freshly collected blood were prepared by allowing the blood to clot for 15 min at room temperature undisturbed, then centrifuged at 2,000×g for 10 min at 4°C. Feces (200 mg) were collected from the anus of the mice, then suspended in 400 μL of PBS with 0.01 M EDTA–Na2. The feces suspension was incubated overnight at 4°C, then centrifuged. The pellet was discarded, and the supernatants were stored at −70°C.
The levels of cytokines, including tumor necrosis factor (TNF)-α, interferon (IFN)-γ, and interleukin (IL)-10 in cell culture supernatant, and IL-4, IL-6, and IL-10 in mice sera was detected with enzyme-linked immunosorbent assay (ELISA) kits as per manufacturer’s instructions (R&D Systems). A standard curve was created to calculate cytokine concentrations using seven step 2-fold dilutions. The antibody response was evaluated by measuring the production of secretory immunoglobulin (Ig)-A and IgG following our previous protocol [38]. Briefly, wells of a 96-well plate were coated with 100 µL recombinant PEDV M epitopes expressed in E. coli (3 µg/mL final concentration) and incubated overnight at 4°C. The plates were then blocked with 3% BSA at 37°C for 1 h. After washing with PBST, 100 µL of immunized mice serum (1:200 diluted) was added to the wells then incubated for 1 h at 37°C. HRP-conjugated goat anti-mouse IgA or IgG antibody (dilution 1:10000 at 37°C, Invitrogen Corporation, Carlsbad, CA, USA) was used to detect titers of IgA and IgG followed by the addition of 3, 3’, 5, 5’-tetramethylbenzidine (TMB). Sulfuric acid (0.5 N) was added to each well to stop the reaction. The plate was immediately measured using an ELISA plate reader (SpectraMax M2e; Molecular Devices, San Jose, CA, USA) at an O.D. of 450 nm.
Assays were performed in triplicate. All results are reported as mean ± SD . Statistical significance was calculated using one-way ANOVA followed with Tukey’s post-hoc test or Kruskal-Wallis followed with Dunn’s post-hoc test in GraphPad Prism (v.8.4.2), whichever is appropriate. Statistical significance was achieved for all analyses with a p-value less than 0.05.
RESULTS
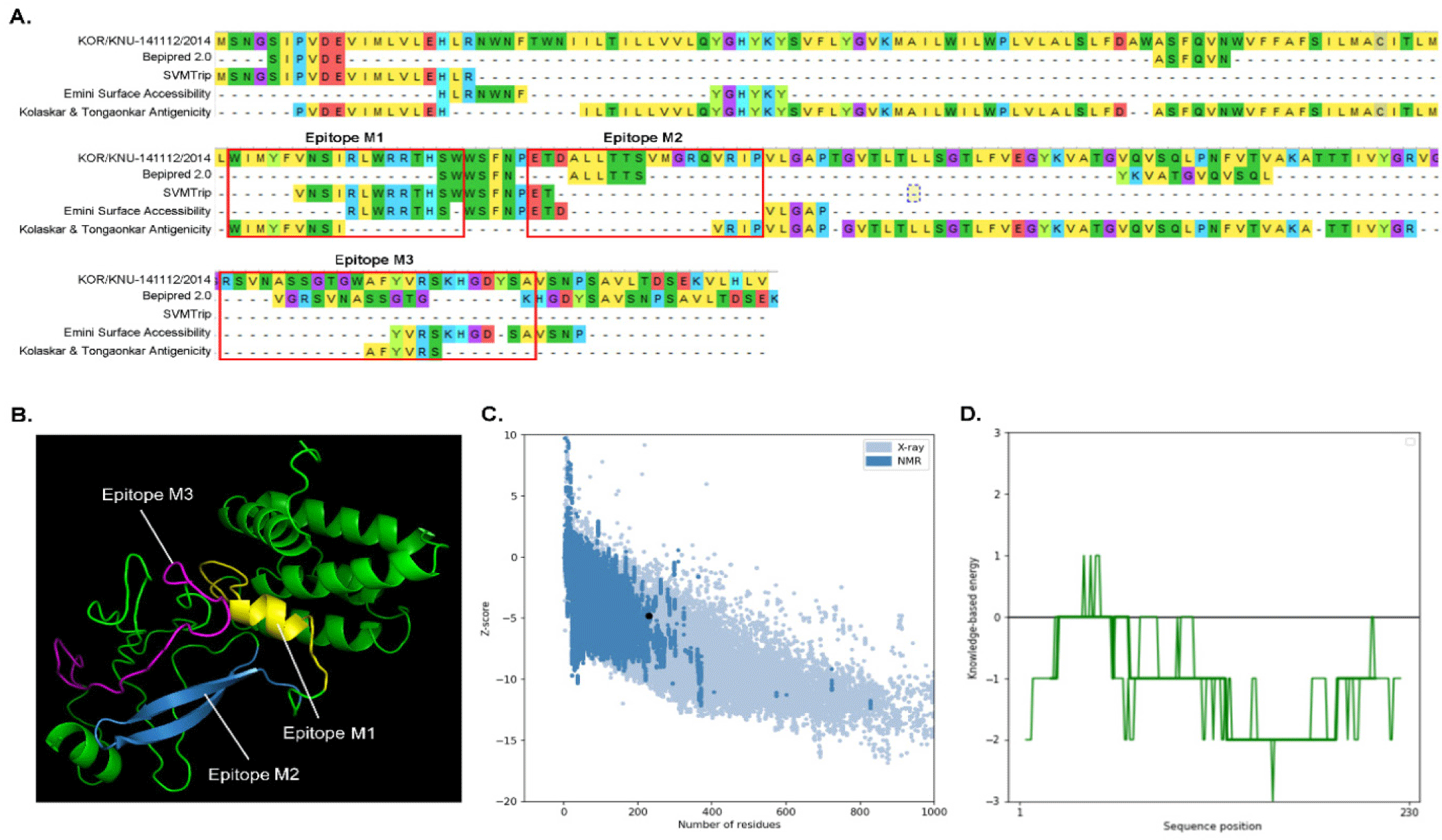
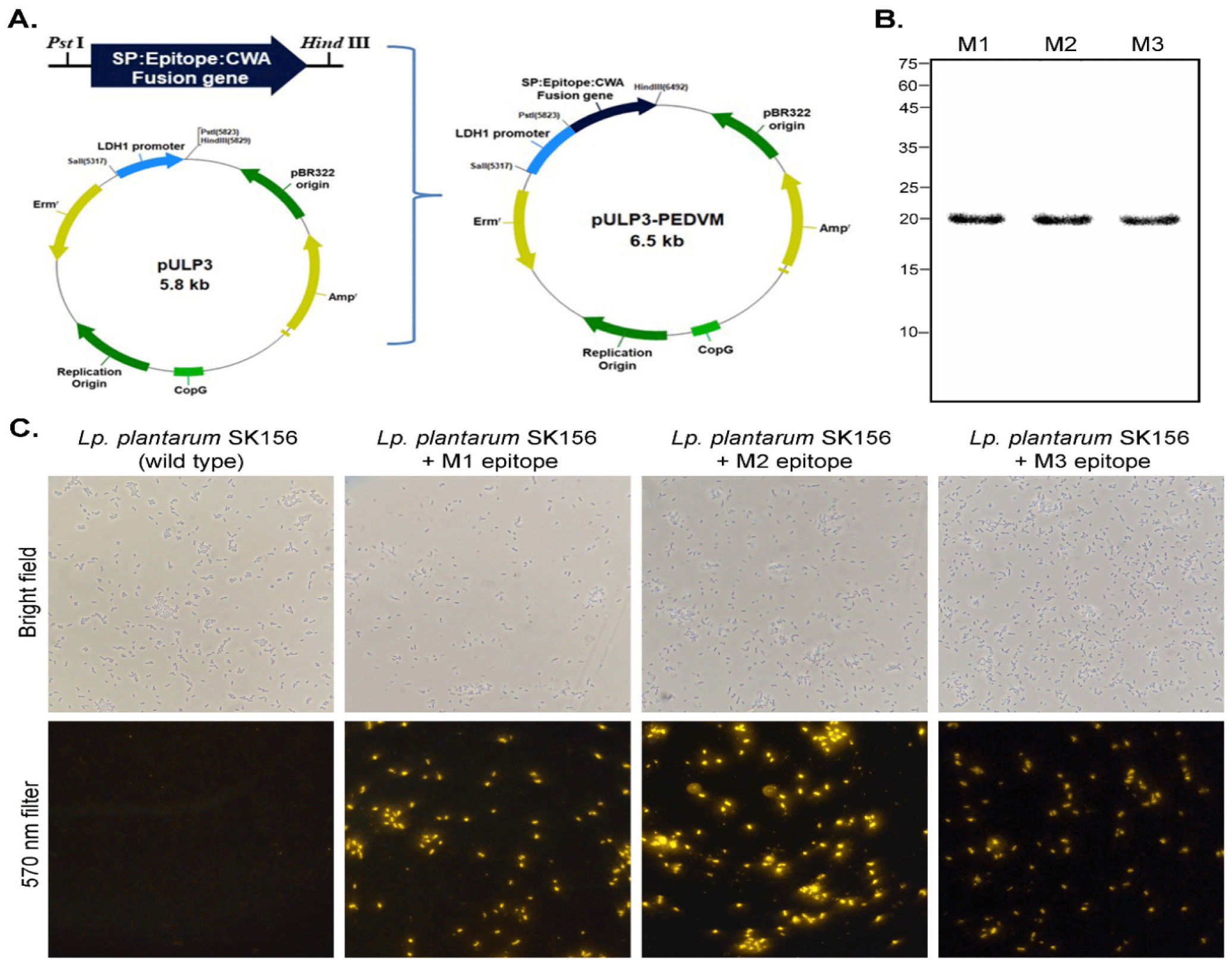
Alignment of M proteins from different PEDV strains showed that the M protein from PEDV strains from Korea (KOR/KNU-141112/2014) has 98.67% and 97.35% similarity to the PEDV strains from Belgium and China, and Japan, respectively (Fig. 1). Conserved regions among these M proteins were considered for B cell epitope prediction. Using Bepipred 2.0 and SVMTrip, several sequences from the M protein of KOR/KNU-141112/2014 strain were predicted. Surface accessibility and epitope antigenicity were also determined to further select candidate epitopes for the surface display experiment. Among the predicted sequences, three candidates were selected and designated as M1 (WIMWYVNSIRLWRRTHSWW), M2 (ETDALLTTSVMGRQVRIPVL), and M3 (RSVNASSGTGWAFYVRSKHGDYSA) (Fig. 2A). The localization of each predicted epitopes within the M protein are shown in Fig. 2B. The predicted structure of the M protein, along with the epitopes was evaluated using ProSA web tool, shows comparability among other PEDV M proteins (Figs. 2C and 2D). The cell surface display vector using SP and CWA of the SlpA of Lb. acidophilus ATCC 4356 as an anchor was constructed by introducing the genes encoding M1, M2, and M3 into the plasmid pULP3, as shown in Fig. 3A, and then transforming Lp. plantarum SK156 via electroporation. To confirm the expression of fusion genes containing M epitopes, proteins were overexpressed in E. coli and western blotting was performed. As shown in Fig. 3B, SP-M epitope-CWA fusion proteins with a combined size of approximately 20 kDa were successfully expressed in E. coli and detected by western blotting. An immunofluorescence assay was performed to determine the cellular localization of M epitopes in the Lactobacillus host. Fig. 3C shows the successful expression and display of all the three M epitopes on the surface of Lp. plantarum SK156. In contrast, wild-type Lp. plantarum SK156 did not exhibit fluorescence, confirming the absence of epitopes of interest on its surface. In addition, brighter fluorescence was observed in Lp. plantarum SK156 expressing the M2 epitope compared with the M1 and M3 epitopes, suggesting that the expression efficiency might differ according to the gene of interest.

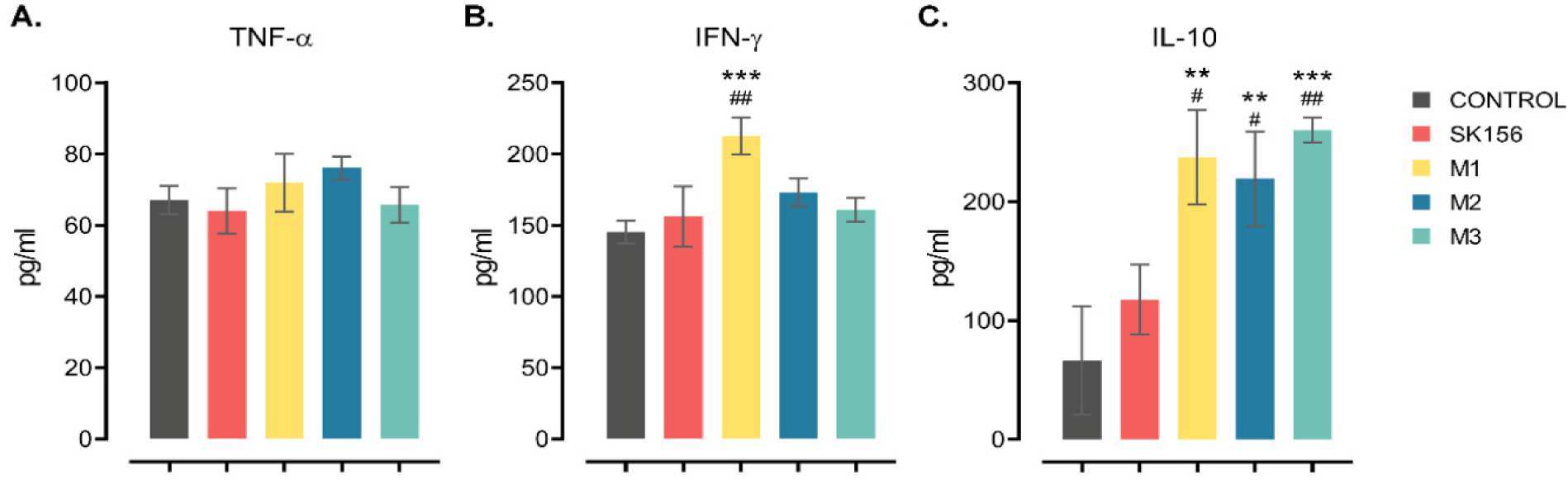
Production of pro-inflammatory and anti-inflammatory cytokines following co-incubation of recombinant Lp. plantarum displaying M epitopes with IPEC-J2 cells was used to assess the type of immune response elicited by the antigen (Fig. 4). Co-incubation of Lp. plantarum displaying M epitopes had no notable effect of the production of TNF-α, regardless of the epitopes. In contrast, Lp. plantarum displaying the M1 epitope induced high level production of IFN-γ (p < 0.05), whereas M2 or M3 epitopes had no significant effect when compared to the control or the wild-type strain. Interestingly, cells co-incubated with Lp. plantarum displaying M epitopes showed a significant increase in IL-10 production compared to cells co-incubated with the control or wild-type Lp. plantarum (p < 0.05). This indicates that Lp. plantarum displaying the M1 phenotype is immunogenic and can elicit both pro- and anti-inflammatory immune responses.
The immunogenicity of the PEDV M epitope surface displayed on Lp. plantarum SK١٥٦ in BALB/c mice was determined by oral immunization (Fig. 5A). The production of antigen-specific antibodies upon immunization was evaluated by ELISA (Figs. 5B and 5C). On day ٢١, M١ and M٢ showed elevated production of fecal sIgA (p < ٠.٠٥). On day ٣٥, mice immunized with Lp. plantarum displaying M١, M٢, and M٣ epitopes exhibited higher production of sIgA than that of the control and wild-type groups (p < ٠.٠٥). In contrast, the mice immunized with Lp. plantarum, indicated that M١ had higher serum IgG levels (p < ٠.٠٥) than that of the other groups. On day ٣٥, the M١ and M٢ groups had higher serum IgG levels than the control of wild-type group (p < ٠.٥). These results showed that epitope M١ was consistent with mounting significant fecal sIgA and IgG production starting on day ٢١ and increasing until day ٣٥ post-immunization.
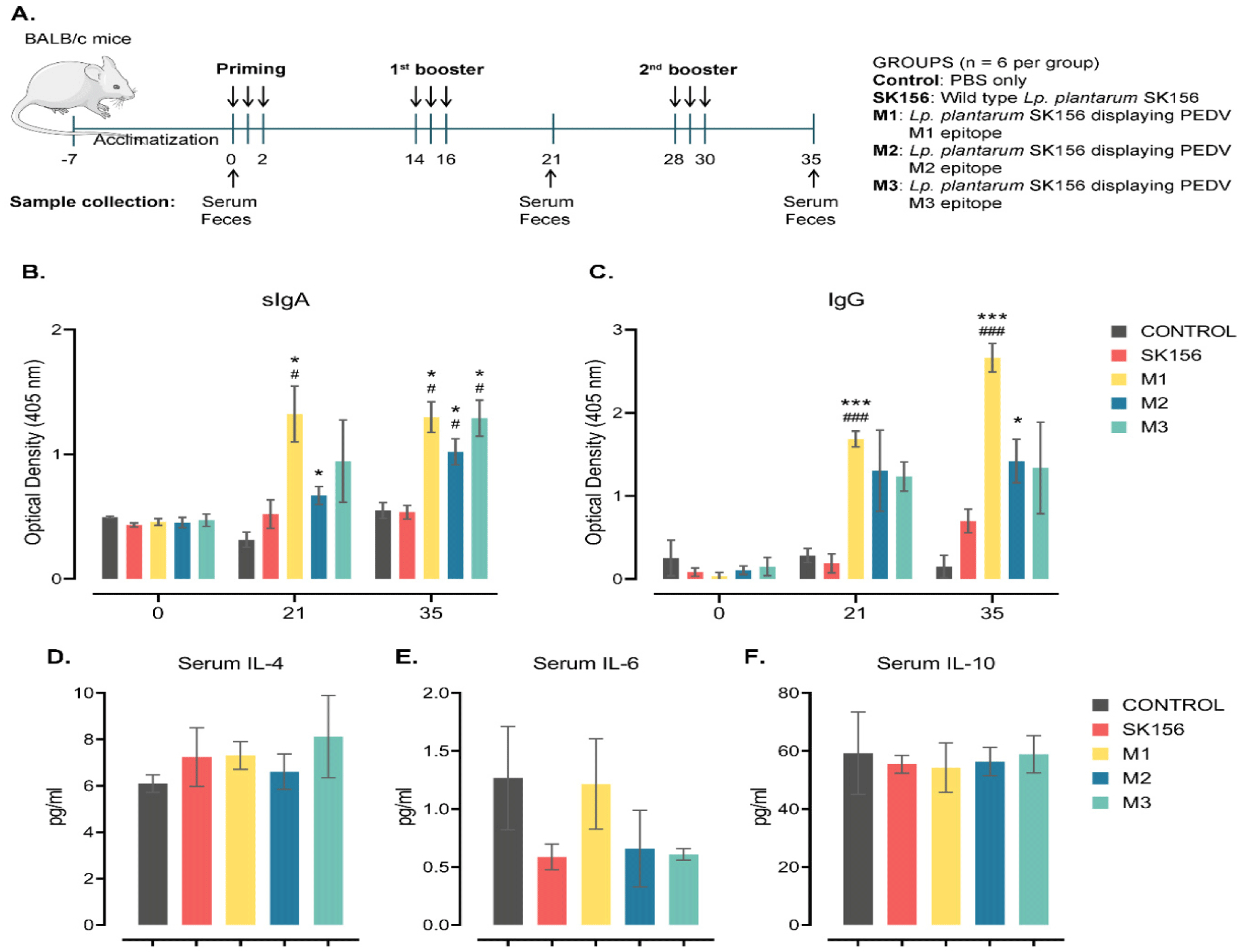
Changes in serum cytokine levels of orally administered recombinant Lp. plantarum SK١٥٦ expressing M epitopes were analyzed using ELISA (Figs. 5D, ٥E, and ٥F). Although marginal changes were observed, serum IL-٤, IL-٦, and IL-١٠ concentrations were not affected by oral immunization with the surface-displayed M epitopes (p > ٠.٠٥).
DISCUSSION
PEDV can be transmitted directly through ingestion of contaminated feces or vomit, or indirectly via inhalation of aerosolized PEDV particles [1,2]. Infection is initiated in the mucosal lining of the nasal cavity, where dendritic cells transfer PEDV particles to CD3+ T cells [1,40]. CD3+ T cells carrying viral particles travel to the intestine through the bloodstream [40]. Thereafter, PEDV invades and multiplies in the intestinal mucosa, such as the villous epithelial cells in the small intestine and jejunum, as well as the surface epithelial cells in the cecum and colon [1,2,41]. For the viral attachment and entry into target cells, the S protein recognizes porcine aminopeptidase N (pAPN), a cellular receptor ubiquitous in small intestinal enterocytes, kidneys, and liver cells [42]. Upon infection, villous epithelial cells are destroyed, damaging the intestine and resulting in acute diarrhea and fatalities in piglets [41]. Current vaccination strategies include the use of whole virions, either live-attenuated or inactivated. Subunit vaccines using viral proteins are potential alternatives to whole-virus vaccines. Alternatively, sows can be artificially infected to induce lactogenic immunity if PED vaccines are unavailable [43]. The fate of immunization is also dependent on the route of vaccination; oral vaccination is known to induce enhanced mucosal immunity against enteric viruses compared with systemic injections [44–46]. Regardless of the strategy employed, immunization for PEDV should provide protection against the virus.
The M protein plays an integral role in the viral life cycle. During the viral life cycle, M protein interacts with other structural proteins and is a key protein in the assembly of viral particles and virion budding [47,48]. The M protein also regulates PEDV replication by interacting with various host factors, such as eukaryotic translation initiation factor 3 subunit L (eIF3L), peptidyl-prolyl isomerase D (PPID), and S100 calcium-binding protein A11 (S100A11) [47,49]. The M protein also contributes to the antiviral defense evasion mechanism of PEDV. The host response upon detection of viral particles includes the activation of type I or III IFNs, which act as the first line of defense against viral infection by blocking viral replication and facilitating viral clearance [50]. Porcine enteric viruses, including PEDV, have developed mechanisms to evade and counteract host antiviral responses [51]. The M protein suppresses the production of IFNs, especially IFN-β, thereby interfering with the interferon regulatory factor 3 (IRF3) signaling pathway [2,5,41]. M protein also exhibits antagonistic activity towards IFN regulatory factor 7 (IRF7), which affects type I IFN production [52]. In addition, the M protein hampers the host immune response by inducing cell cycle arrest at the S phase via the cyclin A pathway [53]. Despite its role in PEDV infection, no subunit vaccine has been developed using the M protein. Nevertheless, because of its conservation among different PEDV strains, M protein is a promising candidate for the development of various detection techniques in diagnostic settings [54,55]. Furthermore, the predicted B-cell epitopes of the PEDV M protein have potential applications in the development of epitope-based vaccines [9,10]. Thus, in this study, three putative B-cell linear epitopes of the PEDV M protein were selected. Using a SP and CWA protein (slpA) from Lb. acidophilus ATCC 4356 [36], M epitopes were displayed on Lp. plantarum SK156. The surface expression and display of M epitopes were evaluated. Immunofluorescence microscopy confirmed successful surface localization of the PEDV M epitopes on Lp. plantarum, verifying the anchorage of the proteins to the cell wall. Hwang et al. [38] successfully displayed SARS-CoV-2 membrane protein epitopes on the surface of Lp. plantarum SK156. In addition, the intensity of immunofluorescence on the cell surface showed that the expression levels of PEDV M epitopes exhibited discrepancies among epitopes, suggesting the possibility of differences in expression levels. These results are in line with observations made when the PEDV epitope was displayed in yeast cells [56]. To the best of our knowledge, this is the first report of PEDV M protein epitopes being successfully displayed on the surface of LAB.
To verify whether the expressed putative epitopes were immunoreactive, the cytokine production in IPEC-J2 cells was measured. PEDV epitopes based on M genes induced higher secretion of IFN-γ than that in the control. In addition, IPEC-J2 co-incubated with epitope M1 secreted elevated levels of IL-10. IFN-γ is a proinflammatory cytokine known for its important function in both innate and adaptive immunity against intracellular infections and tumor suppresion [57]. IFN-γ enhances antigen processing and presentation, increases leukocyte trafficking, triggers an anti-viral milieu, improves anti-microbial functions, and influences cell growth and cell death. [57–59]. During viral infections, IFN-γ interferes with viral replication and interacts with the viral receptor, resulting in the suppression of virus replication [57,60,61]. It also has been shown that production of IFN-γ after immunization is associated with better immune response against viral infections [62]. Recently, Liu et al. [63] reported that surface-displayed porcine IFN-λ3 in Lp. plantarum inhibits PEDV infection in IPEC-J2 cells. This suggests that a stronger IFN-γ response could correlate with higher survival rates in PEDV-infected pigs, similar to what has been observed in other diseases [58,60,62,64].In contrast, IL-10 is an anti-inflammatory cytokine, with key immunoregulatory function during viral and microbial infections [65]. IL-10 counteracts the excessive inflammation caused by Th1 and CD8+ T cell activities and acts as a signal for hyperinflammation [65,66]. IL-10 plays a crucial role in maintaining a balance between pro-inflammatory and anti-inflammatory immune responses, that is, the efficient eradication of pathogens and avoidance of harmful immune responses to infections [66]. Thus, a balance induction of both IFN-γ and IL-10 is necessary for a more effective immunization.
Vaccination aims to stimulate the generation of neutralizing antibodies. Several studies have highlighted the importance of humoral and mucosal responses to PEDV vaccination [44,45,67,68]. In the current study, we observed high levels of PEDV-specific sIgA and IgG in mice immunized with PEDV M epitopes on days 21 and 35, most notably epitope M1. Secretory IgA is an essential effector molecule that neutralizes exogenous antigens [20]. It is produced mostly in the intestinal mucosa, but has been found to be also dominant in the colostrum and milk [46]. IgG plays an important role in systemic viral clearance and is found in the serum and colostrum [46]. Through the gut-mammary axis, PEDV-specific sIgA produced during immunization can be passed from the sow to the piglets via the sow’s milk, supporting the piglet’s immunity against PEDV (also known as lactogenic immunity) [44,46]. Lactogenic immunity is important for inhibiting PEDV replication in the intestines and preventing clinical diseases in piglets. In addition, induction of higher levels of IgA and IgG has been correlated with proper production of neutralizing antibodies [67,69,70]. Although most studies have focused on the S protein of PEDV, the results of the present study are consistent with these data. Oral or intranasal inoculation with recombinant Lb. casei expressing PEDV S protein in pregnant sows and mice results in high levels of IgA and IgG [23]. Li et al. [30] have demonstrated that Lb. casei expressing PEDV S protein induced higher levels of IgA and IgG production in mice. In another study, mice that were orally administered PEDV S1 and S2 protein-expressing Lb. acidophilus had high levels of anti-PEDV-specific IgG and IgA antibodies [29]. Immunization with Lb. johnsonii carrying core-neutralizing epitopes of the S protein resulted in high levels of IgA and IgG in pregnant sows and maternal milk, indicating that immunity can be transferred to piglets [25]. This indicates that, similar to other studies, Lp. plantarum expressing the PEDV M epitope effectively induces the production of sufficient protective antibodies against PEDV infection.
However, we observed negligible changes in the cytokine profiles of the mice immunized with Lp. plantarum expressing the PEDV M epitopes. This is contradictory to the IPEC-J2 results in this study, where high IFN-γ and IL-10 levels were observed. In other studies using PEDV-S protein, immunization led to higher levels of IL-4, IL-6, and IL-10 [25,29,30]. Several factors can be attributed to the observations in this study, such as the type of vaccine or epitope used, dose, timing of measurement, and the specific cytokines being measured [71]. Thus, further careful examination of the effects of immunization using surface-displayed PEDV M epitopes on cellular immune responses is necessary.
CONCLUSION
In this study, a surface display system for the heterologous expression of PEDV M epitopes on Lp. plantarum was constructed and the display of the M epitopes was successfully demonstrated. Moreover, Lp. plantarum displaying the M1 epitope elicited elevated production of IFN-γ and IL-10 in IPEC-J2 cells and high levels of antigen-specific antibodies in mice. The results of the present study highlight the application of surface display in lactobacilli as a potential antigen delivery vector, and the capability of the PEDV M protein as an immunogen to develop candidate vaccines for PEDV. Understanding the protective capability of this response during a challenge is an interesting approach for future research.