
ANNOUNCEMENT
Probiotics are known to contribute to animal intestinal health, performance, and productivity [1]. They are widely used as a food additive in the animal industry. When selecting a probiotic strain, the strain should have acid/bile salt tolerance and cell-adhesion abilities for intestinal survival, as well as other functional properties, including immunomodulatory, antimicrobial, and antioxidant abilities [2,3].
Ligilactobacillus ruminis is an anaerobic, gram-positive bacteria that is autochthonous in the gastrointestinal tract of many animals. It is a lactic acid bacteria found in the large intestine of swine [4]. L. ruminis reportedly has an immunomodulatory effect and can suppress pathogens in the host [3]. However, few studies have documented the potential probiotics properties of L. ruminis. In this study, the genome of L. ruminis CACC881 was analyzed for its potential probiotic properties, and a comparative genomic analysis was performed on four other L. ruminis strains. Fecal samples from 30 days old weaned pigs (Duroc × Landrace × Yorkshire) were collected from a pig farm in Jeongeup City in North Jeolla Province, Korea. The samples were serially diluted with sterile saline buffer and cultured on de Man, Rogosa, and Sharpe (MRS; Merck KGaA) medium under anaerobic conditions at 37°C for 24 h. After randomly selecting single colonies, they were transferred onto MRS medium for further culturing. Identification was conducted through 16S rRNA sequencing, employing the primers 518F (5’-CCAGCAGCCGCGGTAATAC-3’) and 805R (5’-GACTACCAGGGTATCTAATC-3’). After identifying L. ruminis CACC881 (KCTC 25583) from the cultured colony, whole-genome sequencing was conducted. Genomic DNA was extracted from L. ruminis CACC881 cultured in MRS medium at 37°C for 24 h, using the UltraClean microbial kit (Qiagen), and then sequenced on the PacBio Sequel II platform (Pacific Biosciences) for whole-genome analysis. The sequenced raw data were assembled using PacBio SMRT analysis software (version 2.3.0, Pacific Biosciences) [5]. Protein-coding sequences (CDSs) were predicted using the Prodigal 2.6.2 program built into the EzBioCloud server, and the information was validated using the National Center for Biotechnology Information blast. Additionally, bacteriocin-related genes were identified using the BAGEL 4.0 web software (http://bagel5.molgenrug.nl/). The genes were functionally annotated using clusters of orthologous group (COG)-based EggNOG and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases [5]. The orthologous average nucleotide identity (OrthoANI) value of the CACC881 strain was compared with that of related strains (ATCC25644, ATCC27780, PEL65, and DSM20403). A heatmap of the OrthoANI values was constructed using the OrthoANI Tool on the EzBioCloud server. Pan-genome orthologs (POGs) were analyzed using UBLAST with an E-value threshold of 10−6 [2]. A Venn diagram of the calculated POGs was constructed using the Venn program [6].
The complete genome of L. ruminis CACC881 comprised one circular chromosome (2,107,343 bp) with a GC content of 43.4%, 1,935 predicted CDSs, and 85 non-coding genes (19 rRNA and 66 tRNA genes) (Fig. 1A). In total, 1,790 CDSs (92.5%) were functionally classified into 19 COG categories (Fig. 1B). Most of the known protein-coding genes were associated with replication/recombination/repair (12.0%), amino acid transport and metabolism (8.4%), translation/ribosomal structure/biogenesis (8.0%), carbohydrate transport and metabolism (5.9%), and transcription (5.9%). Among the L. ruminis strains, the complete genome of CACC881 was most similar to that of strains DSM20403 (97.3%) and ATCC25644 (97.3%) (Fig. 1C). The pan-genomes of CACC881 and its related strains contained 2,480 POGs, of which 1,523 (61.4%) were core groups shared by all five strains. In total, 219 POGs (8.8%) were unique to the CACC881 strain, which included 60 genes encoding hypothetical proteins (Table 1). The most abundant COG categories in strain-specific orthologs were replication/recombination/repair (16.0%), cell wall/membrane/envelope biogenesis (5.0%), and transcription (4.6%). The genome of the strain CACC881 encoded eight clustered regularly interspaced short palindromic repeats (CRISPR)-related genes/proteins, two immunomodulatory-related genes (tagF and dltA) [6], two antioxidant genes (bcp and ahpC) [7], and genes related to the biosynthesis of vitamin B groups (ribT, ribF, frdA, ribU, ybjI, and cobC/phpB) [8]. Additionally, Class I bacteriocin-related genes were observed, including those related to the regulation of nisin biosynthesis (nisK), nisin immunity (nisI), and lanthipeptide biosynthesis (PSR47_00020) [9]. The findings indicate that the strain CACC881 contained genes related to probiotic characterization, such as tolerance to acid (clpB, queA, and grpE) and bile salts (cbh), and lactate synthesis (ldh) [10]. Notably, genes related to the regulation/immunity of bacteriocin (nisK and nisI) and peroxiredoxin (ahpC) were found only in the genome of strain CACC881, and not in the genomes of the other four L. ruminis strains (Table 2). These findings predict that L. ruminis CACC881 will play a role as potential probiotic including characterization as bacteriocin, biosynthesis of vitamin B group, antioxidant, and immunomodulatory abilities, and possible contributions to gut health and pathogen protection. The complete genome sequence of L. ruminis CACC881 may also contribute to the understanding of probiotic characterization and possible probiotic functions in animals.
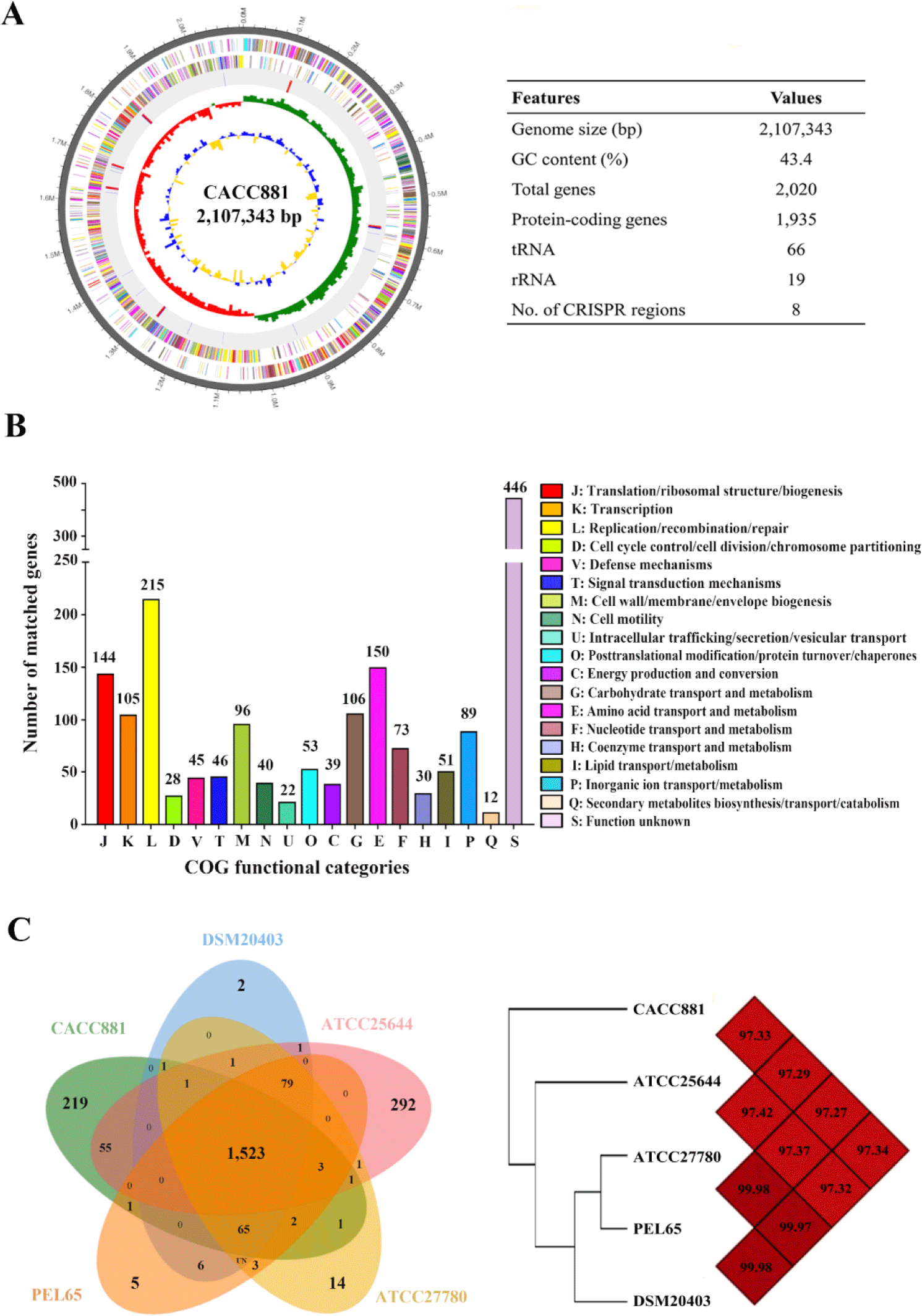
COG, Clusters of Orthologous Groups; POG, pan-genome orthologous group; J, translation, ribosomal structure, and biogenesis; K, transcription; L, replication, recombination, and repair; D, cell cycle control, cell division, and chromosome partitioning; V, defense mechanisms; T, signal transduction mechanisms; M, cell wall/membrane/envelope biogenesis; N, cell motility; U, intracellular trafficking, secretion, and vesicular transport; O, posttranslational modification, protein turnover, chaperones; C, energy production and conversion; G, carbohydrate transport and metabolism; E, amino acid transport and metabolism; F, nucleotide transport and metabolism; H, coenzyme transport and metabolism; I, lipid transport and metabolism; P, inorganic ion transport and metabolism; Q, secondary metabolites biosynthesis, transport, and catabolism; S, function unknown.