
INTRODUCTION
The gut microbiota plays a vital role in supporting the health and development of piglets, particularly during the weaning transition [1]. In the suckling phase, the gut microbial community is altered and influenced by oligosaccharides present in the sow’s milk, which promote the proliferation of beneficial bacteria, including Lactobacillus [2]. These oligosaccharides also support the proliferation of genera such as Escherichia and Streptococcus, which contribute to the development of an anaerobic intestinal environment [3], conducive to colonization by various genera including Bacteroides, Bifidobacterium and Clostridium, thus enriching the diversity of the intestinal microbial community [4]. However, a solid-type weaning diet, which refers to the solid food given during the weaning period, dramatically alters the bacterial communities due to its high proportion of grain and crude protein content [5]. Cao et al. [5] reported that soybean and pectin-rich diets could potentially reduce the proportion of Lactobacillus and increase the relative abundance of Prevotella in the large intestine. Moreover, the abrupt proliferation of Escherichia and Shigella is attributed to the high protein levels in the diet [6]. This disruption of the intestinal microbiota creates an environment susceptible to infection by Proteobacteria, such as Escherichia and Salmonella, which are typical post-weaning diarrhea pathogens [7,8]. Infection-induced inflammation of the intestine also creates a favorable environment for the growth of Proteobacteria [9]. Nitric oxide, generated during the intestinal inflammatory response, is converted into nitrate, which supports the growth of Escherichia coli strains carrying the nitrate reductase gene [10]. Additionally, increased blood flow to the inflamed intestine raises oxygen levels, thus resulting in an increased proportion of facultative anaerobes such as Proteobacteria [11]. This shift disrupts anaerobic conditions and initiates a cycle of adverse conditions which ultimately lead to a loss of bacterial diversity [2]. Therefore, maintaining gut homeostasis during the weaning transition by regulating microbial communities is a crucial challenge in the swine industry.
Probiotic microorganisms have been evaluated as non-antibiotic approaches to restore intestinal microbial balance and inhibit pathogenic microbial infections by producing health-promoting bioactive compounds such as short-chain fatty acids (SCFA), bacteriocins, enzymes, and vitamins [12,13]. Lactiplantibacillus plantarum, a member of the beneficial probiotic group, includes the subspecies Lactiplantibacillus argentoratensis, previously known as Lactobacillus argentoratensis. This bacterium is a gram-positive, facultative anaerobe capable of both homo- and heterofermentation [14]. This bacterium produces SCFA and other metabolites like lactate and acetate, which contribute to gut health by supporting beneficial microbial functions and reducing pathogenic populations through the production of bioactive compounds like hydrogen peroxide. Additionally, the ability of L. argentoratensis to ferment carbohydrates through the Embden-Meyerhoff-Parnas and phosphoketolase pathways enhances its metabolic versatility [15]. In our previous study, we isolated L. argentoratensis AGMB00912 (LA) from the stool of healthy swine and demonstrated its in vitro antimicrobial activity against pathogenic microorganisms, which was primarily mediated by the production of SCFA and improvements in the intestinal microbiota [16]. Building upon these findings, the current pilot-scale study focuses on comparing the gut microbiome of weaned piglets with and without dietary supplementation with LA, aiming to assess its potential impact on the intestinal microbial community structure during the weaning period.
MATERIALS AND METHODS
The LA used for supplementation was isolated in our previous study [16] and inoculated into De Man-Rogosa-Sharpe broth (BD Difco™, Becton Dickenson). The culture was incubated at 37°C for 24 h, followed by centrifugation at 3,264×g for 20 min at 4°C. After centrifugation, the cell pellet was suspended in phosphate-buffered saline 1X (Gibco) and diluted to 1.0 × 108 CFU/mL. The LA was then administered to the piglets in the LA group via oral gavage each day.
Eight 25-day-old castrated male piglets (Landrace × Yorkshire, 5.97 ± 0.43 kg) from the same herd were purchased from a commercial farm. After three days, the piglets were randomly assigned to one of two groups: piglets administered a normal diet for only 10 days (control, n = 4), and piglets administered a normal diet daily supplemented with 1.0 × 108 colony forming units of LA (n = 4). The diet was prepared following the nutritional guidelines outlined in the ‘Korean Feeding Standard for Pigs’ (Table 1). All animal experiments were approved by the Animal Ethics Committee of the National Institute of Animal Science, Korea (approval No. NIAS 2021-503). The experiment was conducted over a total of 10 days, and on the final day (day 10), stool samples (100 g) were collected from each piglet through gentle rectal stimulation. The samples were immediately stored at –80°C until analysis.
Values supplied per kilogram premix feed concentrations: Vitamin A 5,000,000 IU; vitamin E, 1,000 mg; Vitamin B1, 150 mg; Vitamin B2, 300 mg; Vitamin B12, 1,500 mg; niacin amide, 1,500 mg; DL-calcium pantothenate, 1,000 mg; folic acid, 200 mg; Vitamin H, 10 mg; choline chloride, 2,000 mg; min 3,800 mg; zinc, 1,500 mg; iron, 4,000 mg; Cu, 500 mg; I, 250 mg; Co, 100 mg; Mg, 200 mg.
The total DNA was extracted using a DNeasy PowerSoil Pro Kit (Qiagen) from 200 mg of feces collected per sample following the protocol provided by the manufacturer. DNA concentrations were measured using a Victor Nivo (PerkinElmer). A universal primer set targeting the V3–V4 regions (341F–805R) was used to prepare the 16S rRNA gene amplicons [17] using the following polymerase chain reaction (PCR) conditions for the first PCR: 3 min at 95°C for heat activation, followed by 25 cycles of 30 s at 95°C, 30 s at 55°C, and 30 s at 72°C, with a final extension of 5 min at 72°C. The PCR product was purified with AMPure beads (Agencourt Biosciences). After purification, 10 μL of the PCR product was amplified for library construction using NexteraXT Indexed Primer. The second PCR had similar conditions, with 10 cycles. The purified product was quantified by qPCR (KAPA Library Quantification kits for Illumina Sequencing) and qualified using TapeStation D1000 ScreenTape (Agilent Technologies), then sequenced on the MiSeq™ platform (Illumina).
Each amplicon sequence variant (ASV) was analyzed via BLAST+ (v2.9.0) using the NCBI 16S rRNA gene database, assigning taxonomy based on the highest similarity. Hits with query coverage or identity below 85% were discarded. Multiple sequence alignments were performed with MAFFT (v7.475), and a phylogenetic tree was built using FastTreeMP (v2.1.10). Microbial community analyses were carried out through QIIME2 (v1.9) [18] using ASV abundance and taxonomy data. Species diversity and evenness in the microbial communities were calculated using the Shannon and Inverse Simpson indices. Alpha diversity was assessed via rarefaction curves and Chao1 values. Beta diversity, based on weighted and unweighted UniFrac distances, was analyzed to identify variations between comparative groups.
Linear discriminant analysis effect size (LEfSe) analysis was conducted to identify biomarkers with significant differential abundance across groups. The analysis was performed using the microbiomeMarker package (v1.2.1) in R (v4.0.1). Initially, the ASV table, taxonomic classifications, and sample metadata were integrated into a phyloseq object. Statistical significance for the Wilcoxon rank-sum test was set at < 0.05. Normalization was performed using the Counts Per Million method. A Kruskal-Wallis test cut-off of 0.05 was applied to detect features with significant differential abundances.
To infer the metagenome’s functional composition from 16S rRNA gene sequences, the analysis pipeline utilized PICRUSt2. Metadata was loaded from a tab separated values file using the readr package. The ggpicrust2 package was employed to convert the predicted metagenome abundance data into Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway abundance using the ko2kegg_abundance function. Differential abundance analysis across treatment groups was performed using the ALDEx2 method within the ggpicrust2 package’s pathway_daa function. This method provided statistically significant pathways, adjusting for multiple testing using the Benjamini-Hochberg procedure to control the false discovery rate. The top 104 features with the lowest adjusted p-values were selected for further annotation. To elucidate the biological interpretation of the data, KEGG Orthology annotations for these features were obtained. The subset of KEGG pathway abundances corresponding to the top 104 features was extracted for downstream analysis.
Gut microbiota diversity was analyzed using QIIME2 (v1.9). Alpha diversity was assessed with Chao1, Shannon, and Simpson indices, while group differences were evaluated using the Kruskal-Wallis test. Beta diversity significance was assessed via permutational multivariate analysis of variance (PERMANOVA) with the vegan package (v2.6.4) in R, using both unweighted and weighted UniFrac distances. Statistical significance was set at p < 0.05. The LEfSe analysis employed an LDA score cut-off of ≥ 3 to determine the effect size, indicating biologically relevant features. The Kruskal-Wallis and Wilcoxon rank-sum tests were applied with an alpha level of 0.05. Core microbiota analysis was performed with a 20% sample prevalence and 0.2% relative abundance. Predicted metagenomic function differences were analyzed using ALDEx2. Adjusted p-values were calculated using the Benjamini-Hochberg method, with a significance threshold set at p < 0.05.
RESULTS
Sequencing of the 16S rRNA genes from the fecal samples produced high-quality reads across both groups (Table 2, 3). The LA group generated 147,406–161,312 reads per sample, while the control group ranged from 160,070–204,688 reads. The average guanine-cytosine (GC) and adenine-thymine (AT) contents remained consistent across all samples at approximately 53.5% and 46.5%, respectively. After stringent quality control and filtering steps, the final dataset retained 523,362 high-quality, informative reads, with an average of 64,554 reads per sample.
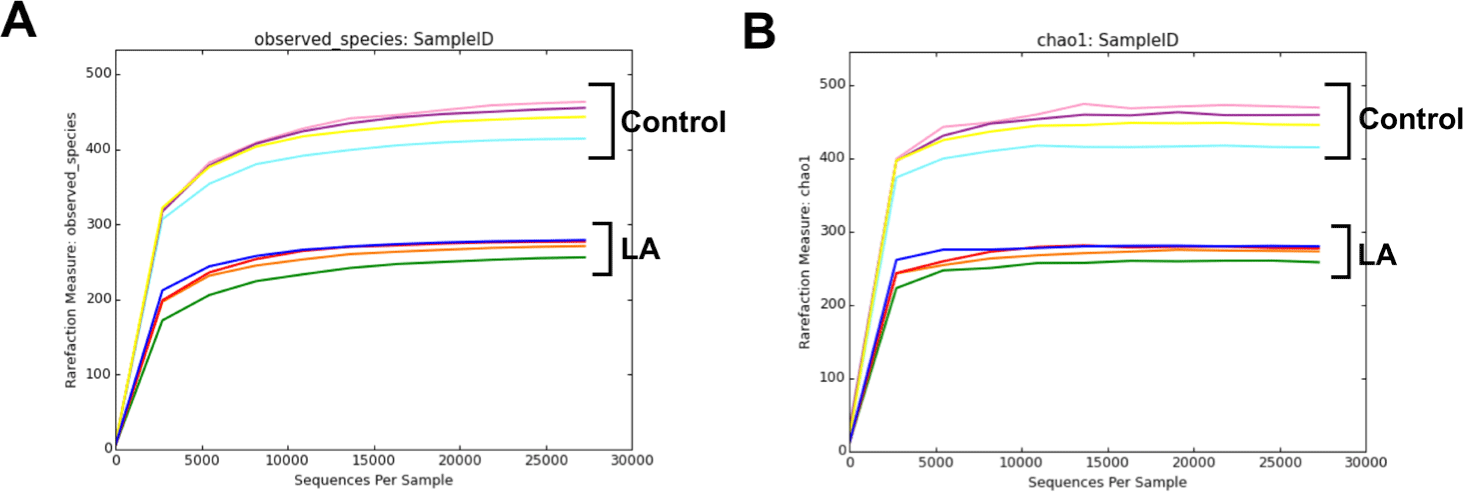
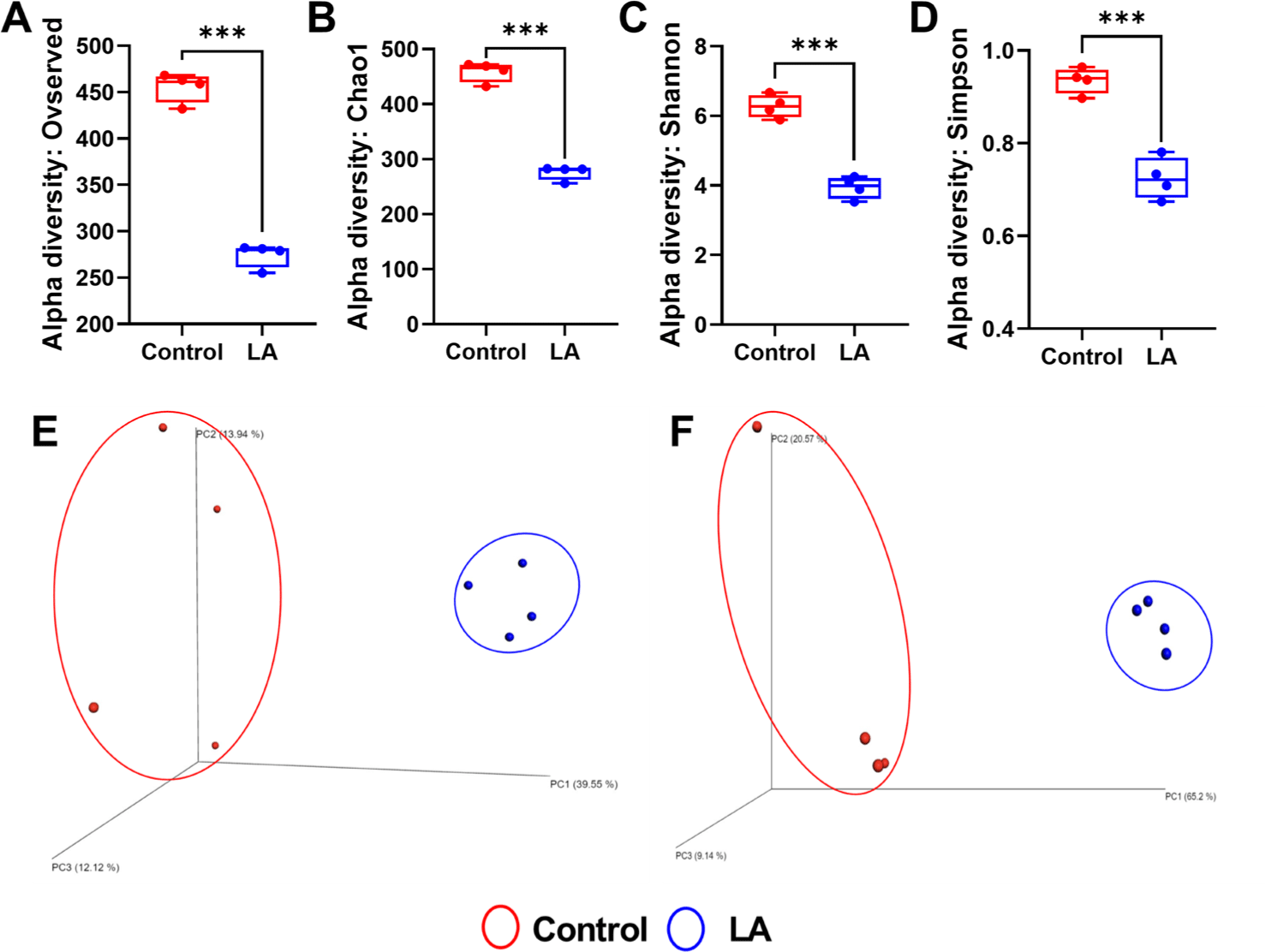
Fig. 1 shows alpha rarefaction curves from 16S rRNA gene sequencing, illustrating species richness in fecal samples from the control and LA groups. Each curve represents a stool sample, with the X-axis showing the number of sequences and the Y-axis showing observed species richness. Stabilization of the curves suggests that the sequencing depth effectively captured microbial diversity, validating the reliability of the subsequent ASV-based analyses. The alpha diversity of bacterial communities in the fecal samples from the weaned piglets was assessed using observed features and Chao1 (indicators of species richness) along with Shannon and Simpson indices (indicating species evenness) (Fig. 2A, 2B, 2C, and 2D). The results showed that the species richness and diversity indices of the control group (observed features: 455.50 ± 16.09, Chao1: 458.91 ± 18.20, Shannon: 6.27 ± 0.33, Simpson: 0.94 ± 0.03) were significantly higher than those of the LA treatment group (observed features: 274.25 ± 92.79, Chao1: 275.73 ± 13.00, Shannon: 3.94 ± 0.31, Simpson: 0.72 ± 0.04) (p < 0.001). The principal coordinates analysis (PCoA) plots, derived from both unweighted (Fig. 2E) and weighted (Fig. 2F) UniFrac distances, demonstrated significant distinctions in the microbial community separation between the control and LA groups in weaned piglets (p = 0.021 and 0.028, respectively). Therefore, dietary supplementation with LA significantly modulates the gut microbiota in weaning pigs.
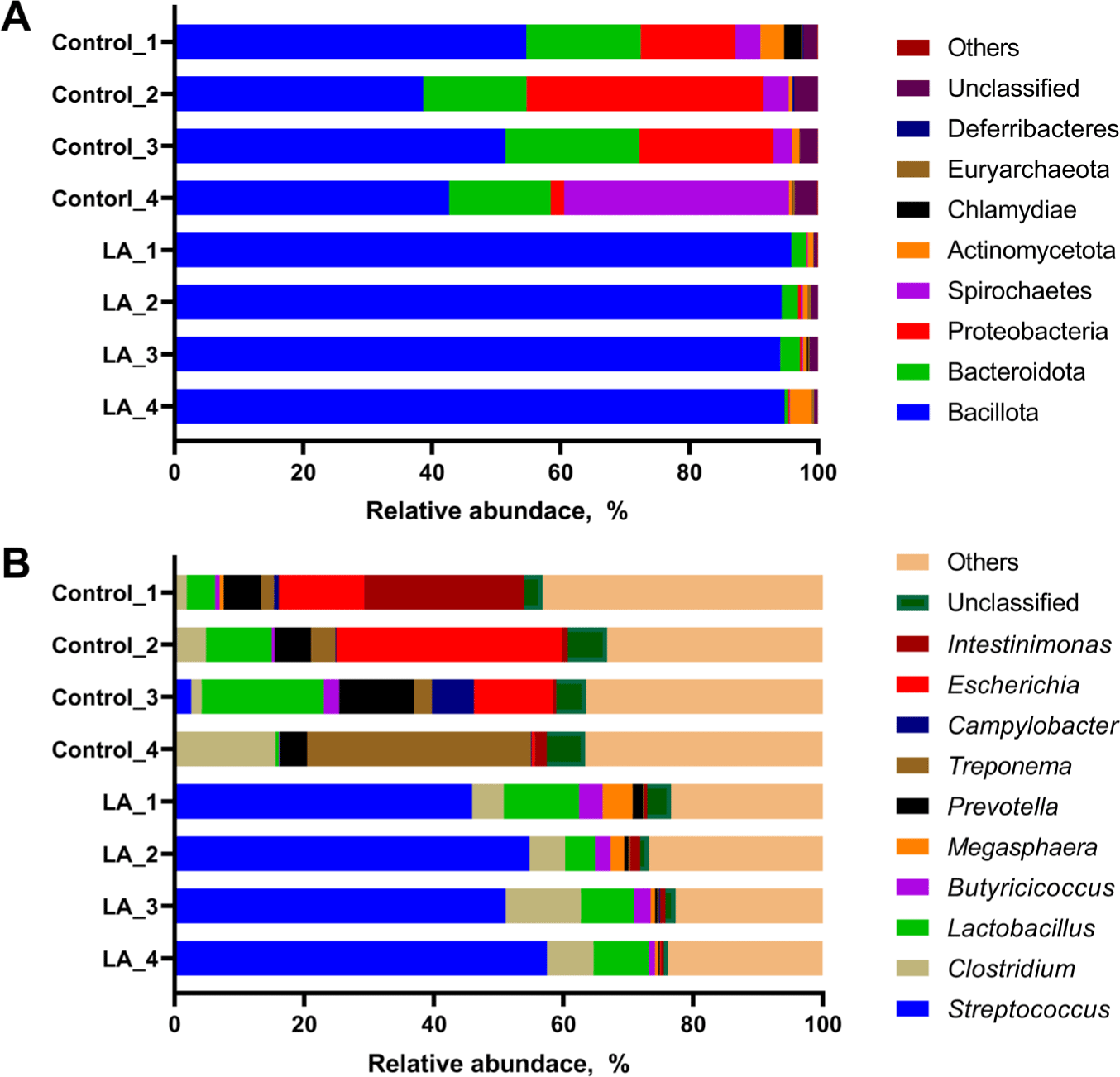
The relative abundance of the microbial populations in LA-treated weaned piglets was analyzed. At the phylum level, the LA group predominantly exhibited Bacillota (94.15%–95.86%) and Bacteroidota (0.56%–3.05%) (Fig. 3A). The control group showed a distribution with Bacillota ranging from 38.68%–54.66% and Bacteroidota from 15.83%–20.85%. Additionally, the relative abundances of Proteobacteria and Spirochaetes in the control group ranged from 2.06%–36.87% and 2.88%–34.91%, respectively. In the LA group, the distributions of these phyla were 0.11%–0.45% and 0.07%–0.28%, respectively (Fig. 3A).
Streptococcus and Clostridium exhibited a relatively high proportion in the LA group, accounting for 45.89%–57.48% and 4.87%–11.65%, while the control group exhibited a microbial community structure characterized by a higher relative abundance of other genera and a lower proportion of Streptococcus and Clostridium (Fig. 3B). The relative abundance of Streptococcus in the control group ranged from 0.12%–2.57%, differing from the distribution observed in the LA group. The relative abundances of the following genera were distributed differently between the control and LA groups: Prevotella (4.14%–11.49% in the control group vs 0.24%–1.59% in the LA group), Treponema (2.03%–34.59% vs 0.07%–0.28%), Campylobacter (0.09%–6.51% vs 0.00%–0.20%), Escherichia (0.49%–34.80% vs 0.00%–0.11%), and Intestinimonas (0.98%–24.70% vs 0.43%–1.49%).
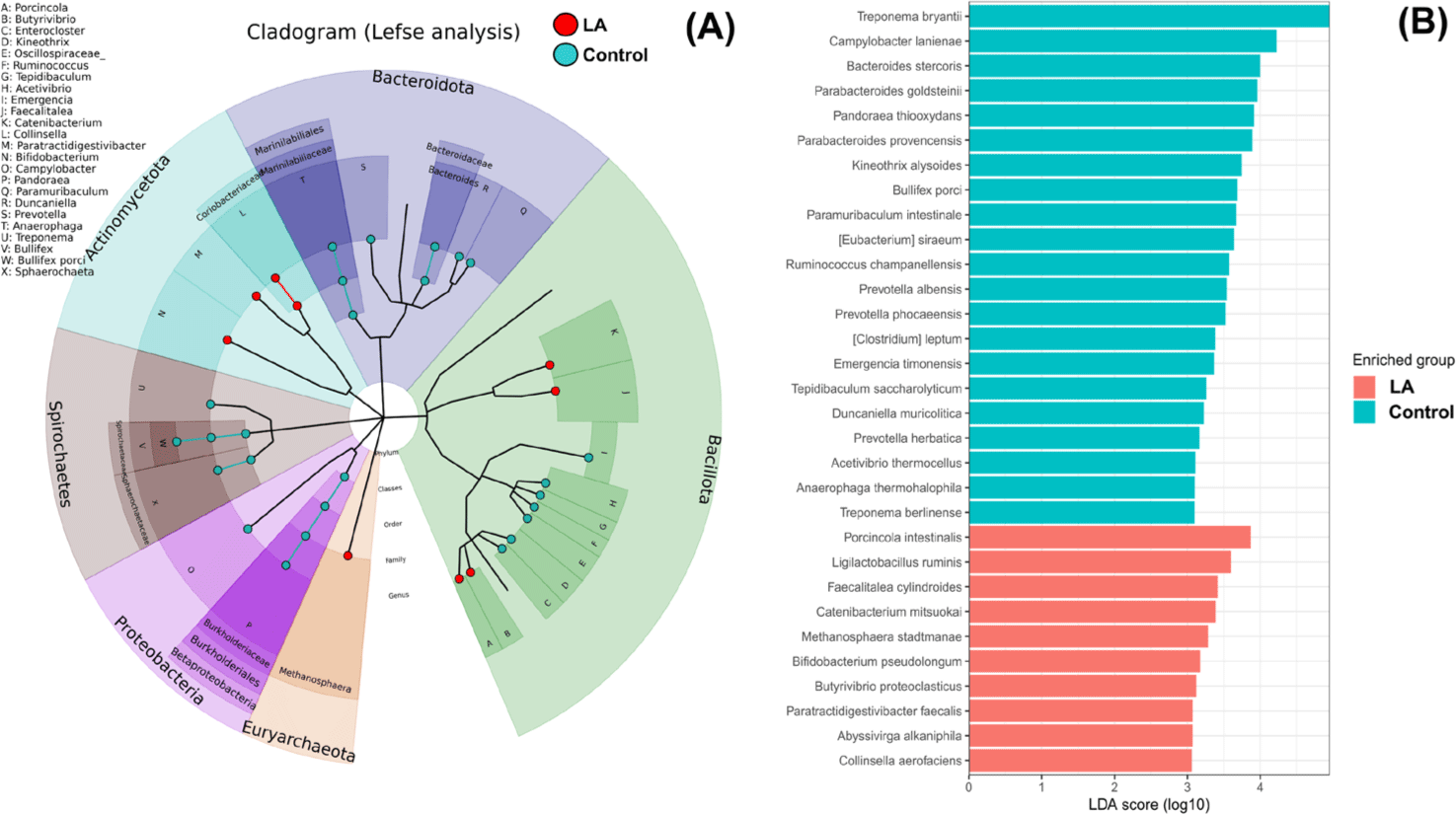
Fig. 4 demonstrates the significantly different taxa in the intestinal microbiota between the control and LA groups. Fig. 4A displays the LEfSe analysis cladogram, spanning from the phylum to genus levels. Fig. 4B presents a histogram displaying the species-level differences in abundance, as indicated by LDA scores > 3. These results revealed the predominant presence of Proteobacteria, Spirochaetes, and Bacteroidota in the control group at the phylum level (Fig. 3). At the genus level, the relative abundance of Treponema, Campylobacter, Bacteroides, and Bullifex was significantly higher in the control group than that in the LA group (p < 0.05). Conversely, LA supplementation significantly increased the proportion of Porcinola, Ligilactobacillus, Faecalitalea, Catenibacterium, Methanosphaera, Bifidobacterium, Butyrivibrio, Abyssivirga, and Collinsella (p < 0.05). To visualize the varying abundance of bacterial genera in the control and LA groups, a hierarchical clustering heat map was generated (Fig. 5). The heat map revealed contrasting relative abundances between the two groups, as indicated by the red boxes.
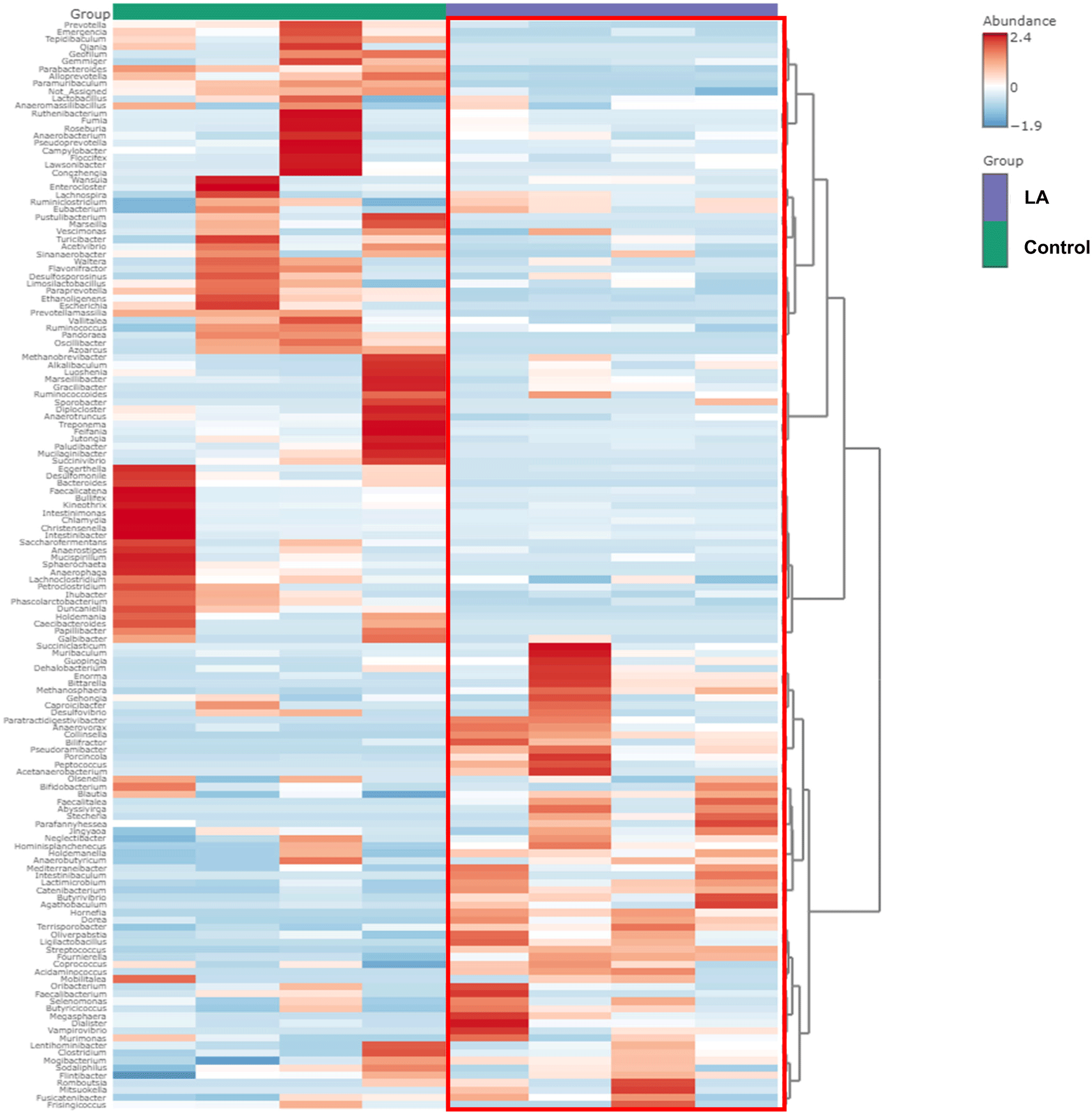
Across all experimental groups, five core bacterial genera were identified, including Streptococcus, Clostridium, Lactobacillus, Prevotella, and Blautia. In contrast, Eubacterium, Escherichia, Intestinimonas, Campylobacter, and Treponema were found only in the control group, with none in the LA group (Fig. 6A and 6B). Therefore, LA supplementation may selectively inhibit pathogenic bacterial genera such as Escherichia and Campylobacter, potentially contributing to a more balanced and beneficial gut microbiota composition in weaned piglets.

To predict the metabolic functions of gut microbiota in weaned piglets treated with LA, PICRUSt2 was used to assess the abundance of KEGG pathways. The heat map depicted the top 22 categories of pathways impacted by the gut microbiota in each group, including cell growth and death, transport and catabolism, membrane transport, signal transduction, folding, sorting and degradation, replication and repair, translation, immune disease, infectious disease, immune system, digestive system, endocrine system, energy metabolism, glycan biosynthesis, terpenoids and polyketides, cofactors and vitamins, carbohydrate metabolism, amino acid metabolism, lipid metabolism, and xenobiotic biodegradation (Fig. 7).
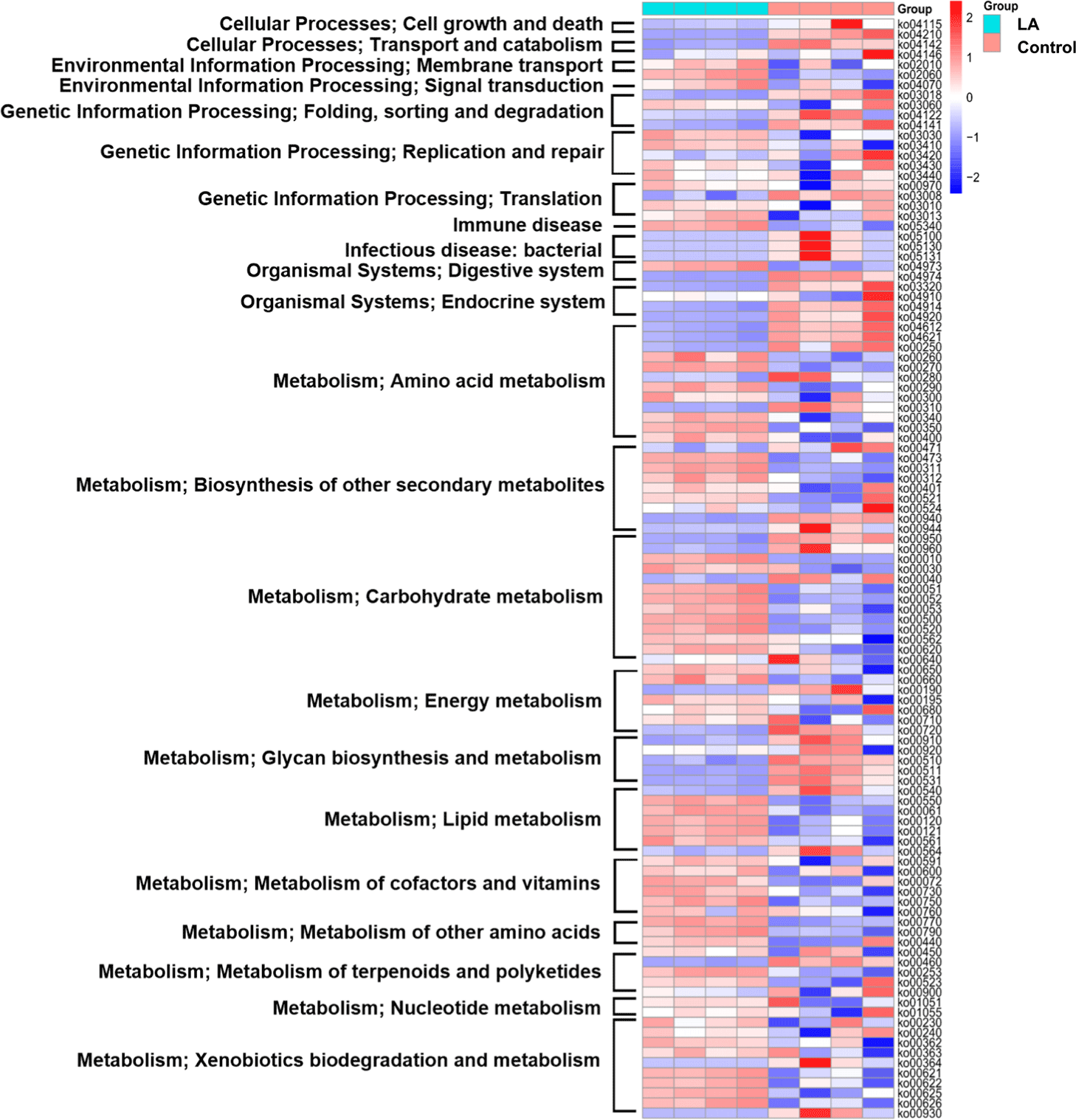
Fig. 8 illustrates the 24 subcategories of metabolic differences. Metabolic activities associated with cell growth and death (specifically the p53 signaling pathway and apoptosis), in addition to transport and catabolism (including lysosome and peroxisome), decreased significantly in the LA group (p < 0.05), compared to those of the control group. While RNA degradation was activated in the control group (p = 0.044), LA supplementation notably alleviated this effect and concurrently activated pathways related to DNA damage repair, including DNA replication, base excision repair, nucleotide excision repair, and mismatch repair. Additionally, the LA group displayed the upregulation of ABC transporter metabolism and the phosphotransferase system (membrane transport), aminoacyl-tRNA biosynthesis, and ribosome (translation), as well as the phosphatidylinositol signaling system. The LA group also exhibited downregulation of the peroxisome proliferator-activated receptors (PPAR) signaling pathway metabolism, while the insulin signaling pathway was upregulated.
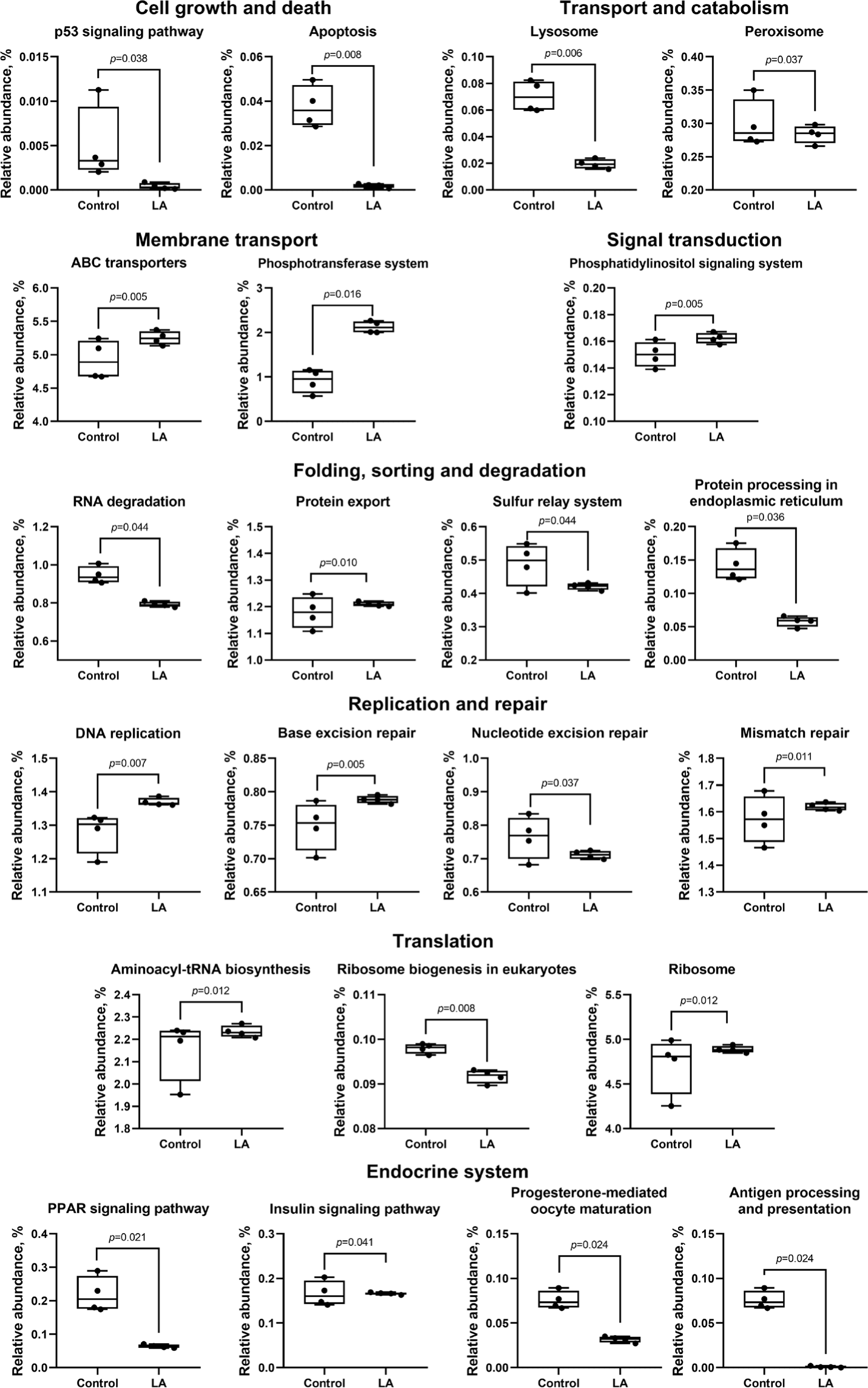
In the control group, metabolism related to bacterial infections was activated, while pathways for bacterial invasion of epithelial cells (p = 0.043), Escherichia coli infection (p = 0.038), and Shigellosis (p = 0.044) were suppressed in the LA group (Fig. 9). Immune system pathways, including antigen processing and nucleotide-binding and oligomerization domain (NOD)-like receptor signaling, were significantly suppressed, while primary immunodeficiency pathways were activated in the LA group.
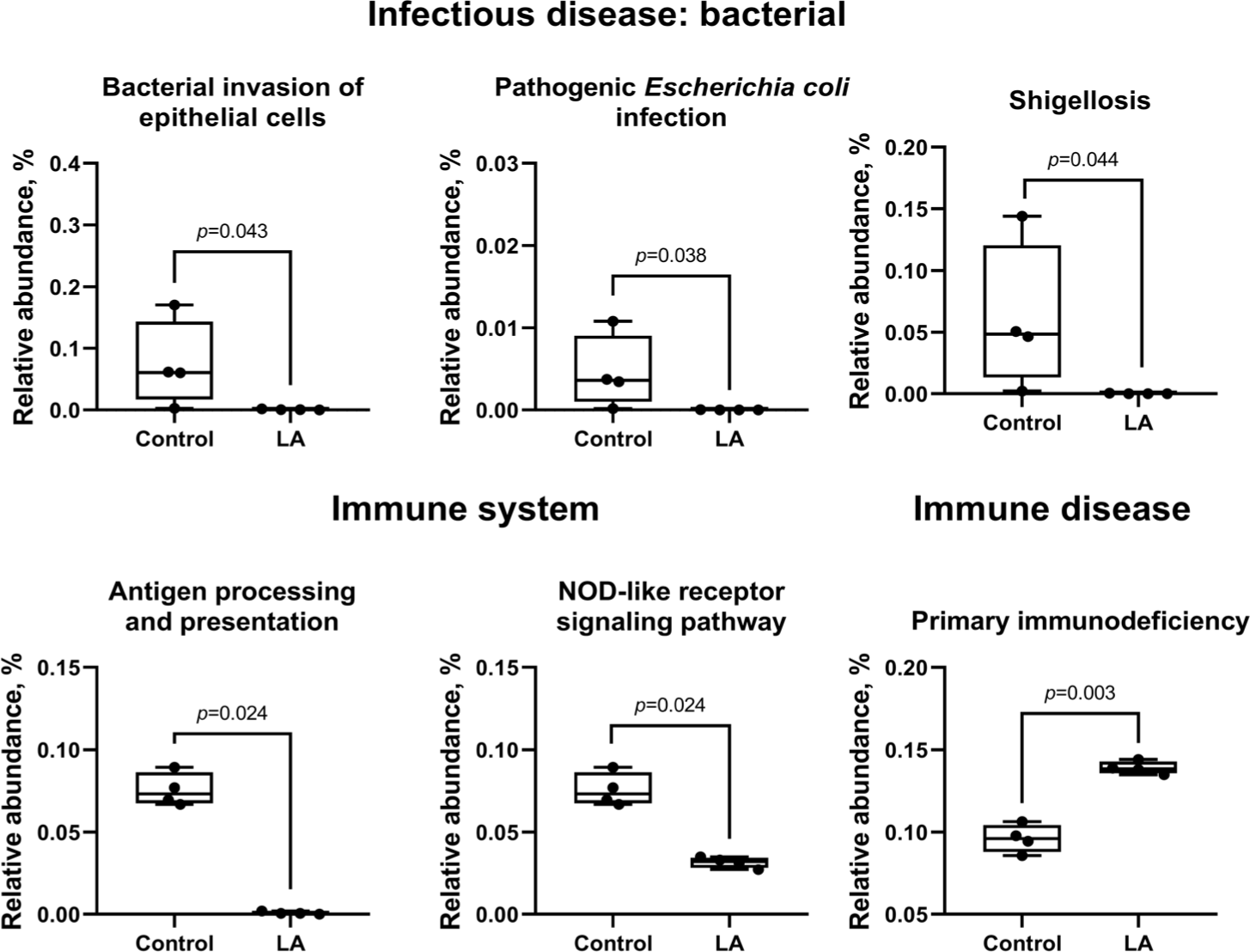
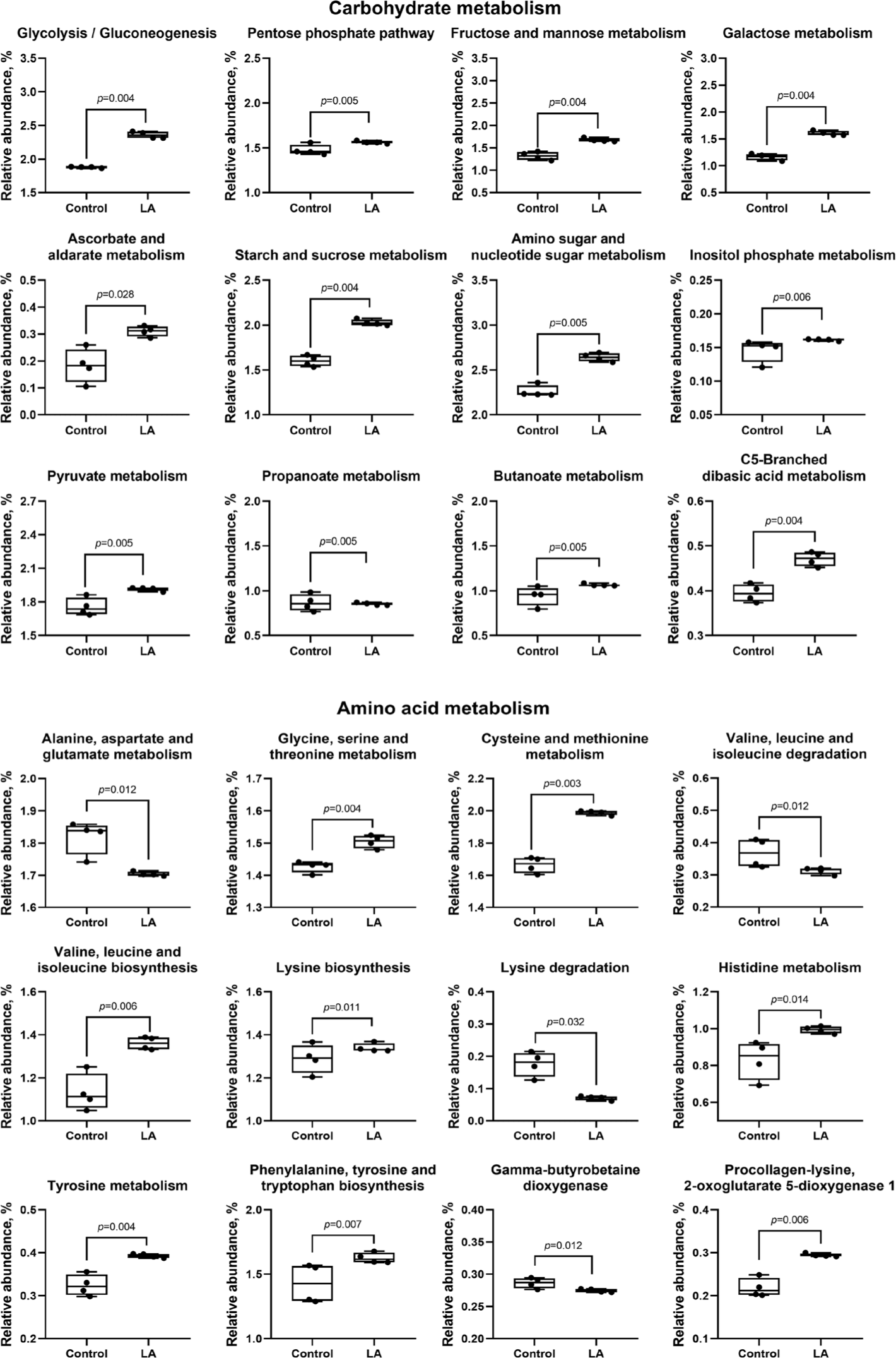
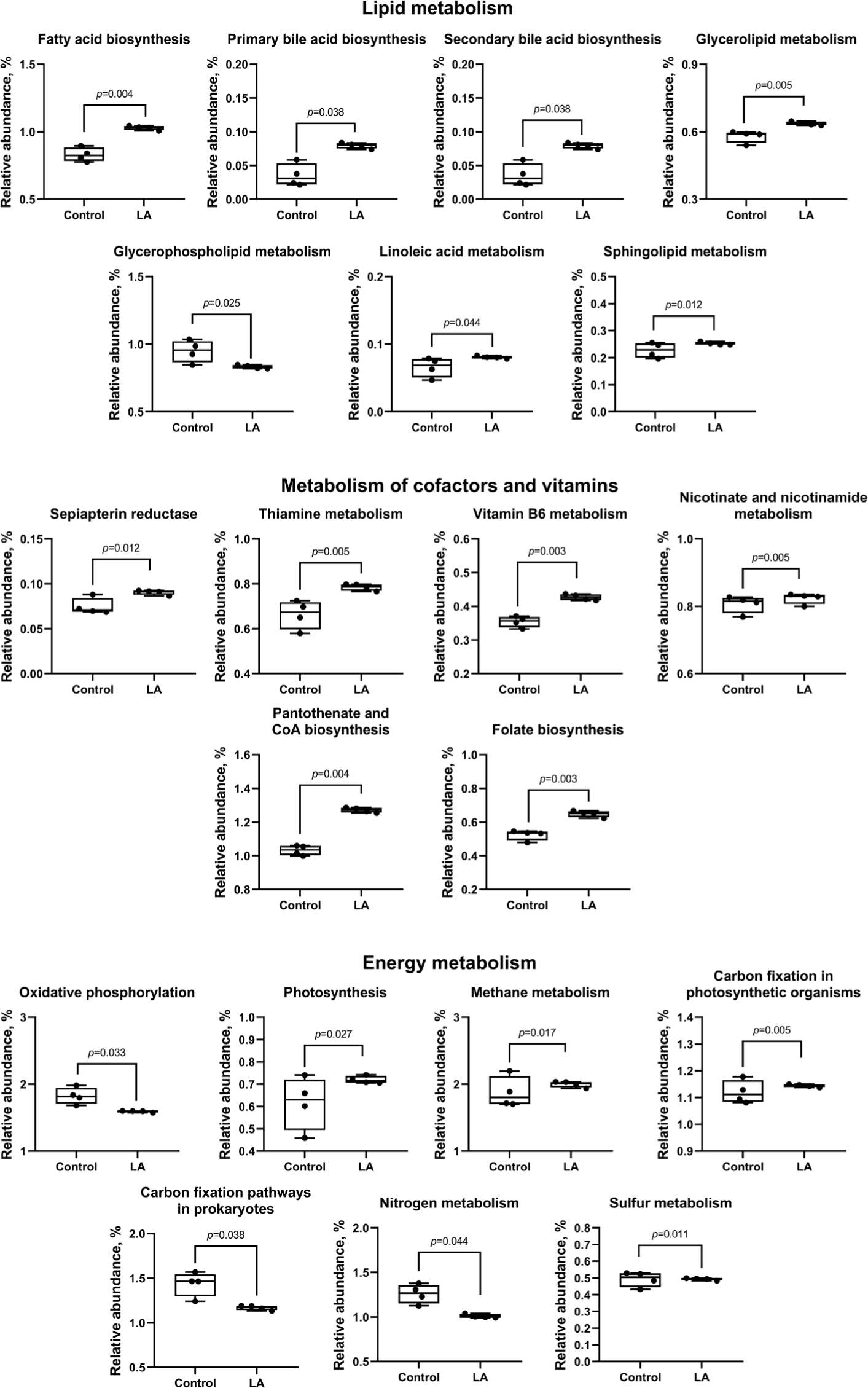
Compared to the control group, LA supplementation upregulated (p < 0.05) carbohydrate metabolism (ascorbate and aldarate, glycolysis, pentose phosphate, fructose, mannose, galactose, starch, sucrose, amino sugar, nucleotide sugar, inositol phosphate, pyruvate, propanoate, butanoate, and C5-branched dibasic acid), as well as amino acid metabolism (glycine, serine, threonine, cysteine, methionine, valine, leucine, isoleucine, lysine, histidine, tyrosine, phenylalanine, and tryptophan) (Fig. 10). In addition, lipid metabolism (fatty acid, primary and secondary bile acid, glycerolipid, linoleic acid, and sphingolipid) and cofactor and vitamin metabolism (sepiapterin reductase, thiamine, vitamin B6, nicotinate, nicotinamide, pantothenate, CoA, and folate) were also increased in LA-treated weaned piglets (Fig. 11). In energy metabolism, oxidative phosphorylation and nitrogen and sulfur metabolisms were suppressed, while photosynthesis, methane production, and carbon fixation in photosynthetic organisms were upregulated in the LA group (p < 0.05).
Analysis of the biosynthesis of terpenoids, polyketides, and other secondary metabolites showed that LA supplementation can significantly increase the biosynthesis of the penicillin, cephalosporin, novobiocin, streptomycin, neomycin, kanamycin, gentamicin, tetracycline, polyketide sugar, ansamycins, and vancomycin groups of antibiotics (p < 0.05). In contrast, LA supplementation downregulated phenylpropanoid, flavone, flavonol, isoquinoline alkaloid, tropane, piperidine, and pyridine alkaloid biosynthesis (Fig. 12). Moreover, LA supplementation mitigated the degradation of other glycans (p = 0.044), enhanced peptidoglycan biosynthesis (p = 0.005), and significantly reduced the synthesis of glycosaminoglycans and lipopolysaccharides (p < 0.05).
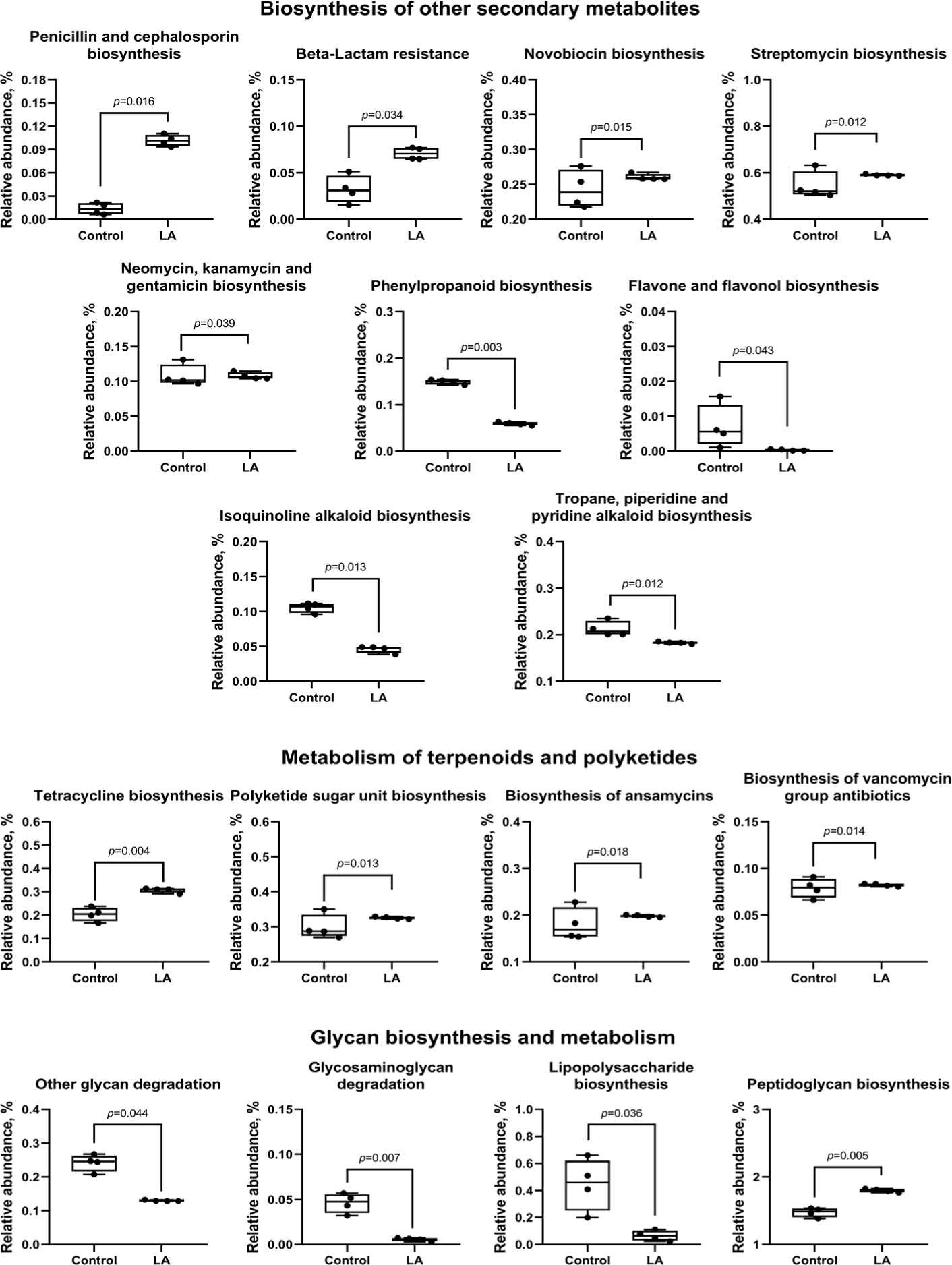
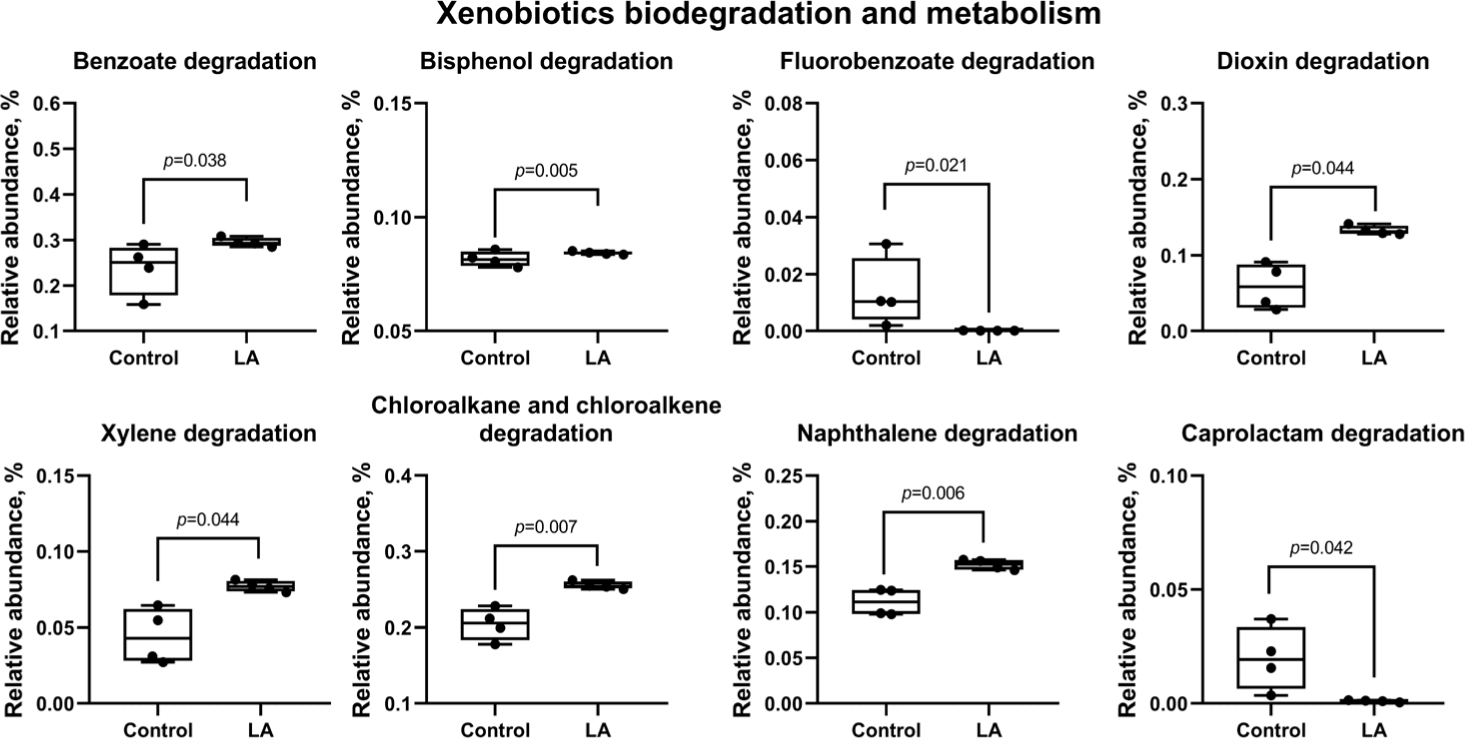
Fig. 13 predicts results for xenobiotic biodegradation and metabolism, indicating a significant reduction in the activation of degradation pathways for benzoate, bisphenol, dioxin, xylene, chloroalkane, chloroalkene, and naphthalene in the LA group (p < 0.05). In addition, the degradation pathways for fluoroacetate and caprolactam were also significantly downregulated in the LA-treated piglets (p = 0.021 and p = 0.042, respectively).
DISCUSSION
During the weaning phase, piglets undergo significant changes in their intestinal microbiome due to separation from the mother sow and sudden dietary changes [2,19]. This study investigated the development of the gut microbiota in weaned piglets, with a focus on the effects of LA, a probiotic strain, on piglet intestinal microbial structure. Our previous pan-genome analysis indicated that LA supplementation produces SCFA, such as lactate, formate, and acetate—key characteristics of LA. Safety evaluations using the virulence factor database and comprehensive antibiotic resistance database confirmed the absence of virulence factors and antibiotic resistance genes, supporting LA as a safe probiotic for commercial use [16]. This study showed that dietary supplementation with LA for 10 days altered the microbial communities in weaned piglets. Alpha diversity, which measures species diversity and richness, is known to fluctuate during the post-weaning period. While some studies report an increase in alpha diversity during weaning [20], others, such as those by Hu et al. [21] and Gresse et al. [2], observed a decrease due to weaning-induced gut dysbiosis. In our study, the control group exhibited significantly higher alpha diversity indices (Observed, Chao1, Shannon, and Simpson) compared to the LA-treated group. A weaning diet typically reduces the proportion of LA while promoting the proliferation of genera such as Prevotella, Bacteroides, Bifidobacterium, Clostridium, Escherichia and Shigella, leading to increased microbial diversity and imbalance [3,4]. These findings suggest that LA supplementation may help stabilize microbial diversity, preventing abrupt changes in the gut microbiome. Furthermore, beta diversity analysis revealed significant differences between the control and LA groups, with each group forming distinct microbial clusters. This pattern indicates that LA supplementation induced a unique microbial structure in the LA group, consistent with previous findings that probiotic strains like Bifidobacterium and Lactobacillus can modulate the gut microbiota in weaned piglets [22,23].
Taxonomic analysis was conducted to further explore the observed differences. The results showed a disruption in microbial balance at both the phylum and genus levels in the control group, while the LA-supplemented group maintained a more consistent microbial structure. Bacillota and Bacteroidota are the two dominant phyla in piglet gut microbiota [21], with Bacillota genera such as Streptococcus, Clostridium, and Lactobacillus playing key roles in producing beneficial SCFA through starch and fiber degradation [24]. The Bacillota to Bacteroidota ratio is an important marker of intestinal community balance and is linked to host health [25]. Generally, an increase in Bacteroidota and a decrease in Bacillota are associated with poor health, as these shifts can affect energy harvest and trigger inflammatory responses [26]. Our findings indicated that LA supplementation enriched Bacillota and preserved the Bacillota to Bacteroidota ratio. Similarly, Guevarra et al. [27] found that supplementation with the probiotic Pediococcus acidilactici modulated this ratio, supporting our results.
Additionally, The LA group had a lower relative abundance of Proteobacteria and Spirochaetes compared to the control group. Proteobacteria, which includes pathogens such as Campylobacter, Escherichia, Helicobacter and Salmonella [21], are often enriched in the intestinal microbiota of piglets suffering from post-weaning diarrhea [28,29]. An increase in Proteobacteria is a common marker of intestinal disorders. Spirochaetes, particularly the genus Treponema, are also known to induce colitis in infected hosts [30]. These findings suggest that LA supplementation may help mitigate microbial imbalances and support gut homeostasis during the weaning transition. Hierarchical clustering analysis further confirmed distinct microbial distributions between the control and LA groups.
To verify the statistical significance of these microbial shifts, LEfSe analysis was performed. Consistent with the taxonomy analysis results, LA treatment significantly reduced the proportion of Proteobacteria and Spirochaetes at the phylum level, and Treponema and Campylobacter at the genus level. Campylobacter is commonly transmitted through the fecal-oral route from sow to piglets, often causing enteritis, especially in piglets deprived of colostrum [31]. Additionally, probiotic treatments have been shown to inhibit the growth of Treponema in weaned piglets, as demonstrated by Zhang et al. [32], supporting the results of this study. Supplementation with LA also significantly increased the relative abundance of beneficial bacteria, including Porcincola, Ligilactobacillus, Faecalitalea, Catenibacterium, Methanosphaera, Bifidobacterium, Butyrivibrio, Abyssivirga, and Collinsella. Porcincola, a gram-positive genus, contains a biosynthetic gene cluster for sactipeptide-like peptides [33], which exhibit antibacterial properties [34]. Ligilactobacillus, formerly part of the Lactobacillus salivarius group, is commonly found in fermented foods and used as a probiotic [35]. It possesses digestive enzymes, produces bacteriocins, and exhibits antioxidant activity [36]. Faecalitalea, a Bacillota member, produces SCFA and has positive effects on insulin secretion and responsiveness [37]. Catenibacterium, a gram-positive anaerobe, synthesizes acetate, lactate, butyrate, and isobutyrate from glucose [38]. Methanosphaera, belonging to the Archaea domain, may improve feed efficiency and reduce methane emissions [39]. Within the phylum Actinobacteria, Bifidobacterium predominates in healthy mammalian intestines and enhances gut health, immunity, and antioxidant activity in weaned piglets [40,41]. Pang et al. [22] also showed that Bifidobacterium promotes growth performance by maintaining gut homeostasis and modulating the intestinal microbiota. Butyrivibrio, an anaerobic butyrate-producing bacterium, was isolated from animal and human intestines [42]. Collinsella, capable of producing SCFAs such as acetate, formate, and lactate, also modulates bile acid and plasma cholesterol levels [43]. Collinsella contains genes for butyrate kinase and phosphate butyryltransferase, suggesting a specialized role in butyrate production [44]. Abyssivirga ferments carbohydrates to enhance nutrient availability and digestibility [45]. In summary, LA supplementation played a crucial role in modulating the gut microbiota of weaned piglets by reducing the abundance of harmful microorganisms such as Campylobacter and Treponema, while enriching SCFA-producing bacteria.
The metabolic functions of microbial communities can be categorized using shotgun metagenomic sequencing. However, this method poses challenges, particularly in the presence of contamination, and is more expensive than 16S rRNA gene sequencing [46,47]. As an alternative, methods like PICRUSt2 have been developed to predict functional profiles based on taxonomic composition. PICRUSt2 predicts metabolic functions from 16S rRNA marker sequences and supports several gene family databases, including KEGG orthologs [46]. In this study, PICRUSt2 was used to predict differentially abundant metabolic functions between the control and LA groups based on 16S rRNA gene sequences. The results revealed distinct expression profiles of the top 22 pathway categories between the two groups (Fig. 7).
One notable pathway, the p53 signaling pathway, is activated in response to intestinal epithelial damage to maintain intestinal integrity. This pathway can lead to cell cycle arrest for DNA repair or trigger apoptosis [48]. Compared to the control, LA supplementation suppressed the p53 signaling pathway, reducing apoptosis and RNA degradation while upregulating DNA repair-related metabolic functions. Additionally, the PPAR-γ signaling pathway, which regulates inflammation in the colon induced by pathogenic bacteria [49], was predicted to be significantly downregulated in the LA group. Lysosome and peroxisome metabolism were also enhanced by LA supplementation, preserving cellular integrity against pathogens [50], maintaining the balance of reactive oxygen species, and protecting cells from oxidative stress [51]. Furthermore, the antigen processing pathway, which prepares antigens for presentation to immune cells, and the NOD-like receptor signaling pathway, which recognizes pathogenic ligands and activates immune responses [52], were modulated by LA treatment. Similar to these findings, Sun et al. [53] reported that Lactobacillus gasseri strain JM1 exerts immunomodulatory effects via the NOD2-mediated signaling pathway. Moreover, the bacterial invasion of epithelial cells, pathogenic Escherichiacoli infection, and Shigellosis were suppressed in the LA group. These findings suggest that LA supplementation can improve fundamental biological processes and enhance systemic health by modulating immune function and disease resistance in weaned piglets.
The weaning transition, marked by the shift from sow milk to solid feed, significantly affects the digestive system and nutrient absorption in piglets [1,4]. In this study, dietary supplementation with LA appeared to upregulate carbohydrate metabolism across 15 distinct pathways compared to the control. Carbohydrates are essential for cellular structure and serve as the primary energy source for living organisms [54]. Carbohydrate metabolism, mediated by the gut microbiota, produces SCFA through processes such as digestion, absorption, and fermentation [55]. The increased proportion of Bacillota genera, including Streptococcus, Clostridium, and Lactobacillus, following LA supplementation, likely enhanced the production of beneficial SCFA via carbohydrate metabolism. The relative abundance of pathways related to amino acid, lipid, cofactor, and vitamin metabolism was significantly increased in the LA group compared to the control. Amino acids like valine, leucine, isoleucine, phenylalanine, and tyrosine are crucial SCFA precursors and fuel the tricarboxylic acid cycle [56]. Additionally, the metabolism of phenylalanine, tyrosine, and tryptophan has been linked to an increase in beneficial bacteria, which helps prevent gut inflammation [57]. Bile acids regulate glucose, lipid, and energy metabolism, while sphingolipids play a role in cell signaling and apoptosis [58]. Linoleic acid, an essential fatty acid, supports cell membrane composition, modulates inflammation, and serves as an energy source for gut epithelial cells [59]. Vitamins B6, pantothenate, and CoA are essential for cellular metabolism, with vitamin B6 playing a key role in fatty acid biosynthesis [60,61].
Intestinal bacteria produce antimicrobial peptides, either ribosomal or non-ribosomal, depending on their biosynthesis pathway [62]. In this study, LA treatment increased the biosynthesis of non-ribosomal peptides, including penicillin, cephalosporin, novobiocin, streptomycin, neomycin, kanamycin, gentamicin, tetracycline, ansamycins, and vancomycin in weaned piglets. These peptides inhibit the growth of pathogenic microorganisms. Additionally, the LA group showed a significant rise in peptidoglycan biosynthesis and a reduction in glycan degradation, indicating a thriving bacterial population that may enhance barrier protection and modulate immune responses, potentially altering the microbiota composition [63,64]. Lipopolysaccharides, from the outer membrane of gram-negative bacteria, are pathogen-associated molecular patterns made of lipid and polysaccharide components [58,65]. The findings of this study suggest that LA supplementation reduced lipopolysaccharide activation in weaned piglets. Furthermore, the activation of degradation pathways for toxic compounds—such as benzoate, bisphenol, dioxin, xylene, chloroalkane, chloroalkene, and naphthalene—was predicted in the LA group, suggesting detoxification of harmful substances in the gut [66,67]. These results indicate that LA supplementation could modulate the gut microbiome of weaned piglets by enhancing antimicrobial peptide production, strengthening gut barrier function, and reducing the risk of endotoxin and toxin exposure.
CONCLUSION
In weaned piglets, dietary supplementation with LA modulated the gut microbiota, suggesting its potential as a valuable additive in the swine industry. Supplementation with LA promoted microbial homeostasis and enhanced metabolic functions of microbial communities, including the biosynthesis of beneficial compounds like antibiotics, glycan structures, and SCFA. These findings suggest that LA may contribute to enhancing immune function and promoting the overall health and growth of weaned piglets. However, this was a preliminary study with a small sample size and only one method of LA administration. Therefore, further large-scale studies are needed to validate these findings and provide a more comprehensive understanding of the gut microbiome’s response to LA supplementation.