
INTRODUCTION
Weaning is a pivotal stage in piglet development, characterized by significant physiological and environmental changes that significantly influence health and productivity [1]. During this period, piglets experience various stressors, including dietary changes, separation from the sow, and adaptation to new environments, which heightens their susceptibility to intestinal disorders and growth impairments [2]. Understanding the biological mechanisms underlying successful weaning adaptation is essential for improving piglet health and welfare in modern swine production systems.
The gut microbiome is crucial for host physiological development and homeostasis, affecting not only intestinal function but also systemic health [3]. Studies show the significant role of gut microbiota in shaping immune development, regulating metabolism, and maintaining barrier function during early life stages [4]. This influence becomes particularly significant during weaning, as dramatic shifts in microbial community composition align with critical developmental changes [5]. The gut microbiota supports host adaptation through various mechanisms, including the production of bioactive metabolites such as short-chain fatty acids (SCFAs), which serve as key mediators of host-microbiome interactions [6].
Host-microbiome interactions are complex biological processes involving diverse regulatory networks and signaling pathways [7]. These interactions are mediated through various molecular mechanisms, including metabolite signaling and immune system modulation [8]. The gut microbiota influences host gene expression patterns across multiple tissues, highlighting its systemic effects on host physiology [9] This interaction is especially critical during weaning, as establishing a stable host-microbiome relationship is key to successful adaptation [10].
The complexity of host-microbiome interactions requires a comprehensive analytical approach that captures multiple layers of biological information simultaneously [11]. Multi-omics integration, a branch of systems biology, offers a powerful tool for understanding complex biological processes [12]. This approach integrates multiple omics platforms, including transcriptomics, metabolomics, and microbiome analysis, to provide a comprehensive view of biological systems [13]. Integrating multiple data types enhances our understanding of the flow of information from microbiome composition to host response, revealing key pathways and mechanisms involved in host-microbiome interactions [14].
This study aims to identify key mechanisms underlying host-microbiome interactions during the weaning transition through multi-omics integration analysis. Furthermore, the critical weaning transition period was examined by comparing samples from the weaning day (W0) and 14 days post-weaning (W14). The integration of microbiome profiling, metabolomic analysis, and host transcriptomics aims to identify the complex relationships between gut microbiota composition, microbial metabolite production, and host physiological responses. The comprehensive approach provides new insights into the molecular mechanisms governing successful weaning adaptation, potentially improving strategies to enhance piglet health and performance during this critical transition period.
METHODS
On the day of weaning, 12 piglets ([Landrace × Yorkshire] × Duroc; initial body weight [BW] = 6.67 ± 0.79 kg; 28 days old; equal male-to-female ratio) were assigned individually to 12 pens. All pigs were fed a mashed diet based on corn and soybean meals to meet their nutritional requirements [15]. Equal-sized pens (dimensions: 232 × 175 cm) were equipped with freely accessible water, feeder, and plastic floor slats. Pigs were housed in temperature-controlled pens with ambient temperatures set between 25°C and 28°C and humidity levels maintained at 50% to 65%. The lighting schedule followed a 12-h light/dark cycle (Fig. 1) [16].
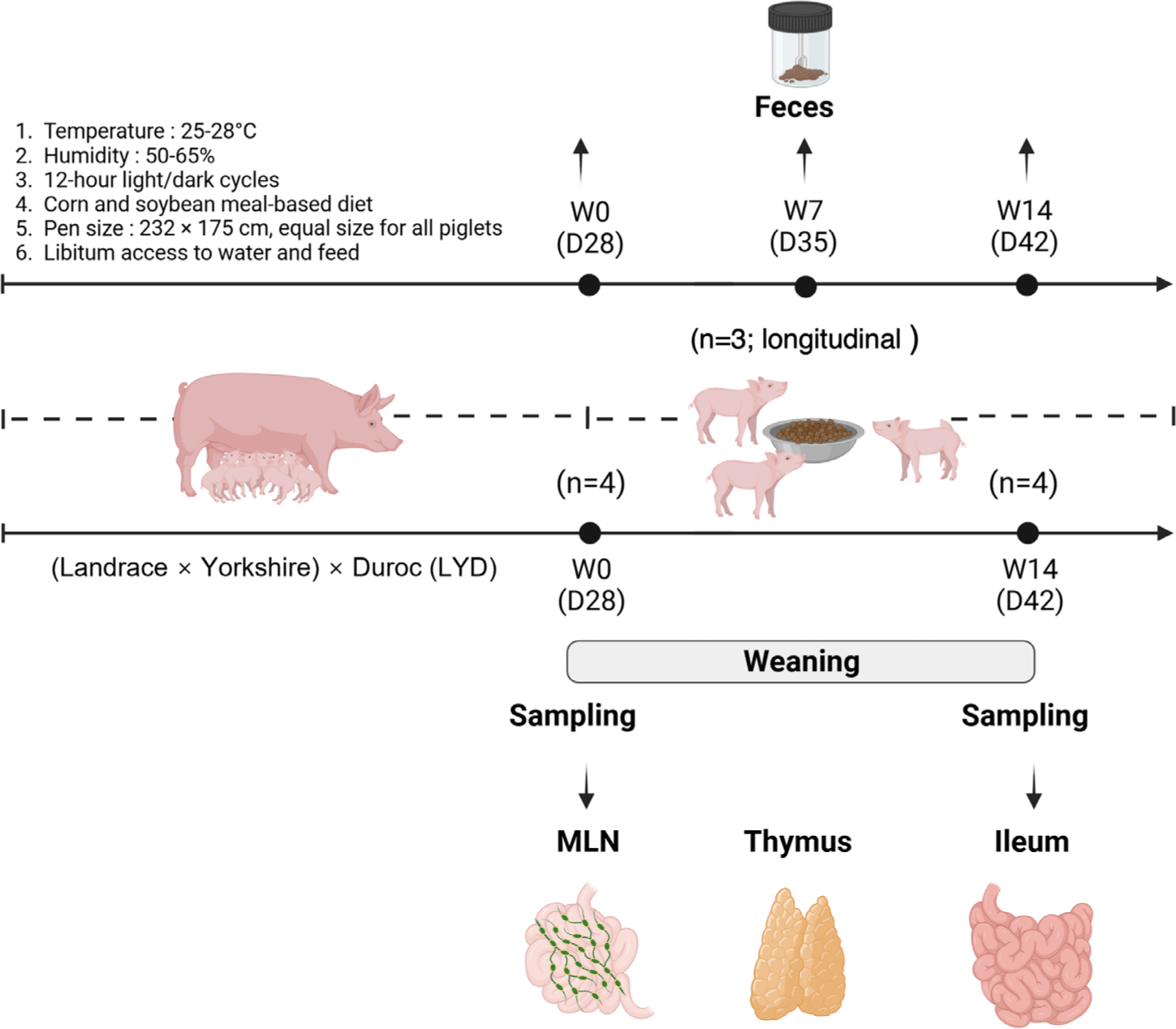
Individual BW was measured and recorded at weaning (day 0, W0) and 14 days post-weaning (day 14, W14). Fecal samples were collected longitudinally from the same six piglets at three time points (W0, W7, and W14; repeated measurements within individuals) and immediately stored at –80°C for subsequent microbiome profiling. After quality control, three longitudinally matched piglets (n = 3) were retained for downstream fecal microbiome analyses. A separate cohort was used for tissue-level analysis. Six piglets were euthanized at W0 and six piglets were euthanized at W14. From these, four biologically independent piglets per group (n = 4 at W0 and W14) were selected for ileal microbiome and integration analyses. Transcriptomic analyses were performed separately on ileum, thymus, and mesenteric lymph nodes (MLNs) collected from the same four piglets per group (n = 4 at W0 and W14). Prior to euthanasia, pigs were anesthetized with a 2 mL intramuscular injection of suxamethonium chloride (Succicholine Inj., BKPharm). Euthanasia was performed by exposure to 90% carbon dioxide with continuous monitoring of vital signs. At each necropsy time point (W0 and W14), the following samples were collected. For microbiome and metabolomic analyses, ileal contents were collected and stored at –80°C. For intestinal morphology analysis, approximately 3 cm segments of ileum were excised, washed with distilled water, and fixed in 50 mL conical tubes containing 10% neutral buffered formalin solution (BBC Biochemical) until microscopic examination. For transcriptomic analysis, tissue samples were collected from ileum, thymus, and MLNs. Ileal tissue segments were gently scraped to remove luminal contents, and all tissues were immediately stabilized in 1.5 mL microtubes containing RNAlater reagent (QIAGEN GmbH), incubated for 24 h at room temperature, and then stored at –80°C until RNA extraction.
Sequencing libraries were prepared to amplify the V3 and V4 regions following the Illumina 16S Metagenomic Sequencing Library protocols. Input gDNA (2 ng) was PCR-amplified using a reaction mix containing 5× reaction buffer, 1 mM dNTP mix, 500 nM of each universal F/R PCR primer, and Herculase II fusion DNA polymerase (Agilent Technologies). The first PCR cycle conditions were: 3 min at 95°C for heat activation, followed by 25 cycles of 30 s at 95°C, 30 s at 55°C, and 30 s at 72°C, ending with a 5-min final extension at 72°C.
The universal primer pair with Illumina adapter overhang sequences employed for the first amplification was as follows: V3-F 5′-GTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG-3′, V4-R 5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAAT CC-3′. The first PCR product was purified using AMPure beads (Agencourt Bioscience). After purification, 2 μl of the first PCR product was amplified using Nextera XT Indexed Primers to construct the final library with the index. The second PCR followed the same conditions as the first but was limited to 10 cycles. The resulting PCR product was then purified using AMPure beads. The final purified product was quantified using qPCR following the qPCR Quantification Protocol Guide (KAPA Library Quantification kits for Illumina Sequencing platforms) and qualified using the TapeStation D1000 ScreenTape (Agilent Technologies). Paired-end sequencing (2 × 300 bp) was performed on the MiSeq™ platform (Illumina) by Macrogen.
Adapter and primer sequences were removed using Cutadapt v3.7 [17]. Sequence processing was conducted in QIIME2 (v2022.8). Amplicon sequence variants (ASVs) were inferred using DADA2 after quality filtering and chimera removal [18]. Taxonomy was assigned using the q2-feature-classifier plugin with a Naïve Bayes classifier trained on SILVA reference sequences (v138). A rooted phylogenetic tree was constructed for phylogenetic diversity analyses. Alpha diversity metrics (observed ASVs and Shannon index) and beta diversity (weighted UniFrac distance) were calculated after rarefaction to an even sampling depth of 1,218 reads per sample, selected based on rarefaction curves. Beta diversity was visualized by principal coordinates analysis (PCoA), and group differences in community structure were assessed using PERMANOVA with 999 permutations. Longitudinal fecal samples were collected from the same individuals. Time-associated patterns were interpreted without an explicit repeated-measures model. Taxonomic composition was summarized at the genus level, and taxa with < 1% relative abundance were grouped as others for visualization.
Functional prediction of the microbial communities was conducted using PICRUSt2 (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States 2) [19]. The ASV table was normalized by 16S rRNA gene copy numbers, and enzyme commission (EC) numbers and Kyoto Encyclopedia of Genes and Genomes (KEGG) orthologs (KOs) were predicted using the reference database. The predicted KOs were mapped to KEGG pathways for functional annotation. Statistical analysis and visualization of functional pathways between groups were conducted utilizing Statistical Analysis of Taxonomic and Functional Profiles (STAMP) software [20]. Significant differences in predicted KEGG pathways between groups were identified using Welch’s t-test, with pathways having a p-value < 0.05 considered statistically significant. The results were visualized employing extended error bar plots in STAMP to show differences in functional potential between the preweaning and post-weaning groups.
For microscopy, the fixed ileum tissues were implanted in paraffin, cut into thin sections, stained with hematoxylin and eosin (H&E), and sealed on slide glass. Fifteen villi and associated their crypts were selected from the H&E slides by a fluorescence microscope (TE2000, Nikon) and NIS-Elements software (Version, 3.00; NIS Elements, Nikon) to measure villus height, width, area, crypt depth, villus height to crypt depth ratio (VH:CD), and number of goblet cells. Intestinal morphology was analyzed using the GLM procedure of SAS (SAS Institute). The experimental unit was the pig, post-weaning day was a main effect.
Total RNA was extracted from ileum, thymus, and mesenteric lymph node (MLN) tissues using the TRIzol reagent (Invitrogen, Life Technology) according to the instruction of the manufacturer [21]. RNA quantity was measured with a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies). For library preparation, 1 µg of total RNA was processed using the Illumina TruSeq™ RNA Sample Preparation Kit. Sequencing was performed with paired-end (2 × 100 base pair) reads on the Illumina HiSeq 2000 platform (Illumina). During library preparation, mRNA was fragmented into small pieces, and these fragments were used to synthesize first-strand cDNA with reverse transcriptase and random primers. The second-strand cDNA was synthesized using DNA Polymerase I and RNase H, followed by an end-repair process. To establish a quality filtering strategy, the raw read data for each sample were assessed using FastQC v0.11.9 [22]. The reads were trimmed with Trimmomatic v0.39 [23] based on quality results using the parameters SLIDINGWINDOW:4:15 and MINLEN:36, which included adaptor removal. The trimmed reads were re-checked with FastQC and aligned to the reference genome (Sus_scrofa.Sscrofa11.1.109) from the Ensembl genome browser (https://asia.ensembl.org/Sus_scrofa/) using the default options in HISAT2 v2.1.0 program [24]. The mapped reads were sorted and converted to binary format using Samtools v1.9 [25]. The raw gene counts for each library were calculated based on exons in the Sus_scrofa GTF v109 (Ensembl) genomic annotation reference file using featureCounts from the Subread package [26]. Differentially expressed gene (DEG) analysis of the raw counts was performed using the edgeR package v.3.40.2 from Bioconductor [27]. To minimize statistical bias in the DEG analysis, genes with raw counts ≤ 10 in all samples were excluded. Raw counts were normalized using the trimmed mean of M-values (TMM) method [28]. A multidimensional scaling (MDS) analysis was performed using the limma package [29] in R, and the results were visualized with the ggplot2 package [30] to assess sample similarity. DEGs were identified for each group in relation to the weaning transition. The adjusted p-value was calculated using the Benjamini–Hochberg method to control the false discovery rate (FDR) below 0.05. A log2 fold change (FC) threshold of ≥ 1 was applied as the criterion for DEG selection.
Biological processes were annotated using Gene Ontology (GO) terms [31] and KEGG pathways [32] through The Database for Annotation, Visualization, and Integrated Discovery (DAVID) [33]. GO annotations were filtered using the DIRECT option and applied for enrichment analysis with the following criteria: GO terms with p < 0.05 and count ≥ 2 were considered enriched. KEGG annotations were enriched using the same cutoff criteria and presented as –Log10 p-values and fold enrichment. Enriched GO terms were grouped with related terms and visualized as a tree map using REVIGO [34]. The most significant GO terms within each group are shown as representative.
For metabolome analysis, samples were extracted with ice-cold 100% methanol (0.5 mL per 100 µL of sample). The extraction involved three cycles of 1-min vigorous vortexing, with 1-min intervals on ice. Samples were centrifuged at 16,000×g for 5 min at 4°C, and the supernatants were filtered through 0.2 µm PVDF syringe filters. Filtered extracts (400 µL) were dried in a speed vacuum concentrator and stored at –80°C until analysis. Before GC-MS analysis, samples were derivatized under nitrogen using bistrimethyl–silyltrifluoroacetamide. Chromatographic separation was conditioned on a 5% diphenyl/95% dimethyl polysiloxane-fused silica column (20 m × 0.18 mm ID; 0.18-µm film thickness) with helium as the carrier gas. The temperature program increased from 60 to 340°C over 17.5 min. Each sample was spiked with nine internal standards (250 ng each): amylbenzene, 1-phenylhexane, 1-phenyloctane, 1-phenyldecane, 1-phenyldodecane, hexadecylbenzene, octadecylbenzene, tetradecylbenzene, and 2,6-di-tert-butyl-4-methylphenol. Analysis was conducted using a Thermo-Finnigan Trace DSQ fast-scanning single-quadrupole mass spectrometer with electron impact ionization (EI), scanning a mass range of 50–750 m/z.
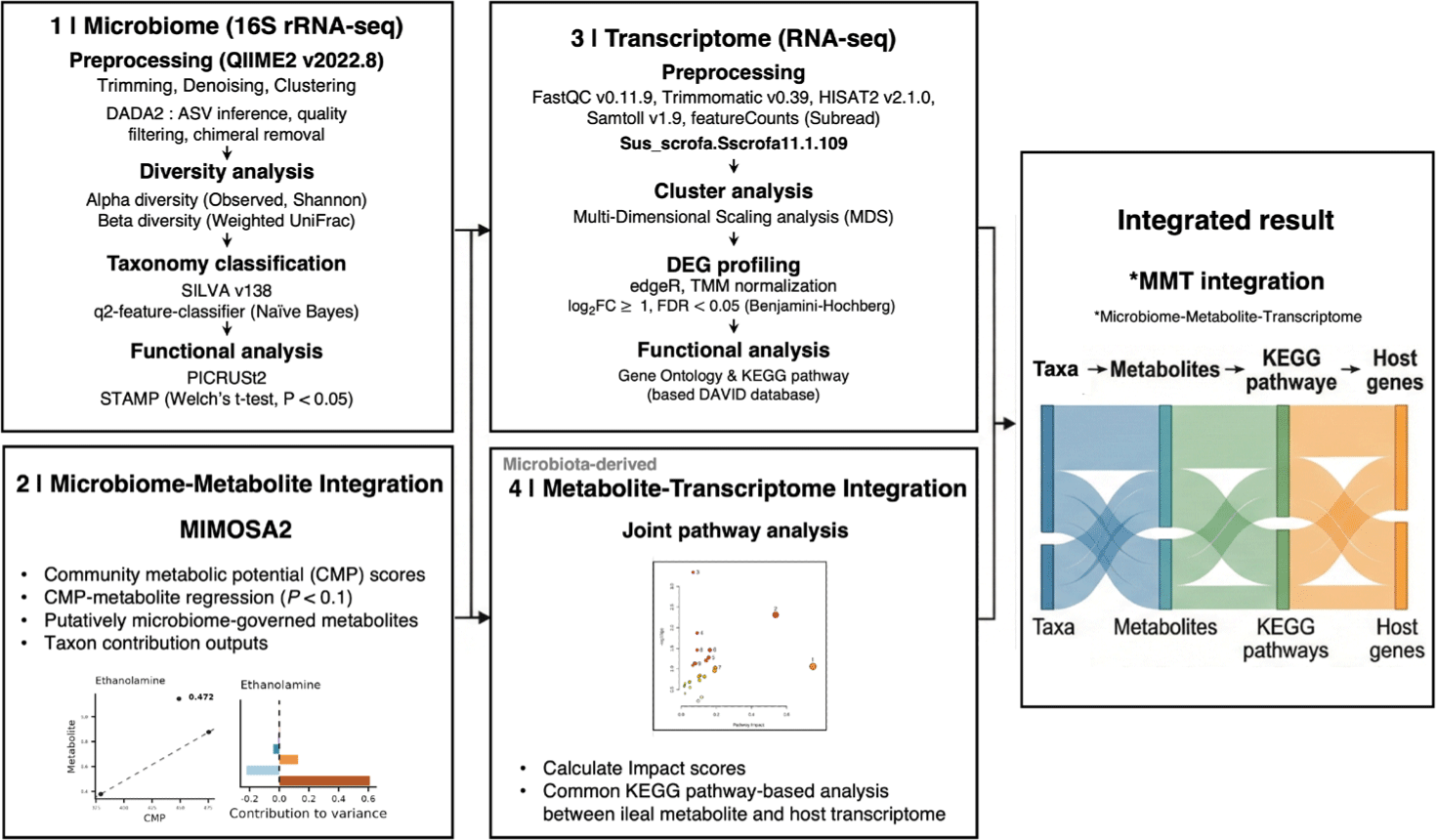
Microbiome and metabolomics datasets were integrated using Model-based Integration of Metabolite Observations and Species Abundances 2 (MIMOSA2) [35]. MIMOSA2 evaluates whether observed metabolite variation across samples is consistent with community metabolic potential (CMP) inferred from reference metabolic reactions and microbial taxonomic abundances. Briefly, MIMOSA2 maps taxa to a KEGG-based reaction reference set to estimate each taxon’s predicted capacity to synthesize and/or utilize specific metabolites. Taxon-level potentials are subsequently aggregated at the community level with weighting by relative abundance, thereby generating CMP scores for each metabolite and sample. Regression models are then applied to assess the relationship between total CMP and measured metabolite levels across samples (p-value < 0.1). Metabolites exhibiting a significant CMP–metabolite relationship under the default MIMOSA2 settings were classified as putatively microbiome-governed metabolites within the model, and multiple-testing adjusted values are reported. In addition, the fraction of metabolite variation explained by the CMP-based model was decomposed into taxon-level contributions to identify putative key contributing taxa. Given that MIMOSA2 provides mechanistically informed evidence derived from a reference metabolic model, resulting inferences were interpreted as hypothesis-generating associations rather than definitive cause–effect relationships. Joint pathway analysis was performed using MetaboAnalyst 6.0 [36] to integrate ileal DEGs and putatively microbiome-governed metabolites identified by MIMOSA2. Metabolite and gene identifiers were mapped to KEGG compound and gene entries using the MetaboAnalyst annotation procedure, and matched features are provided in Supplementary File 1. Pathway enrichment was evaluated by over-representation analysis based on the hypergeometric test, and multiple testing correction was applied using the FDR with results reported in Supplementary File 2. Pathway impact (Topology) was calculated based on degree centrality, and the combine queries option was used for integration. A Sankey diagram was generated using Flourish to visualize multi-omics links across microbial taxa, metabolites, pathways, and DEGs. Microbial features were summarized at the family level. Links between taxa and putatively microbiome-governed metabolites were informed by MIMOSA2 taxon-level contribution outputs. Links between metabolites and KEGG pathways were informed by joint pathway analysis results, including pathway impact, and pathway-linked DEGs were derived from the KEGG annotation used in the enrichment analysis. The workflow shown in Fig. 2 also summarizes the steps used to generate the Sankey diagram, linking microbial taxa, putatively microbiome-governed metabolites, KEGG pathways, and host DEGs.
RESULTS
Analysis of 4,713,808 high-quality sequencing reads from fecal samples revealed distinct microbiome profiles across weaning timepoints (W0, W7, and W14). Rarefaction curves confirmed sequencing depth saturation. Overall, 12,698 unique ASVs were identified, with post-weaning samples showing increased abundance. Alpha diversity metrics indicated significantly higher microbiome complexity after weaning, with greater species richness and Shannon diversity indices in post-weaning piglets (W7, W14) than in day-of-weaning piglets (W0) (Fig. 3A). Beta diversity analysis using weighted UniFrac distances revealed clear temporal clustering patterns between the day-of-weaning and post-weaning piglets, with W7 and W14 samples substantially overlapping but distinctly separated from W0 samples (Fig. 3B). These findings indicate a rapid and sustained shift in gut microbiome composition after weaning.
Taxonomic analysis revealed substantial compositional shifts in the fecal microbiome during weaning (Fig. 3C). At the genus level, Prevotella showed the greatest increase in relative abundance in post-weaning samples, becoming dominant by W14. Lactobacillus abundance initially decreased at W7 but recovered by W14. Microbial diversity further increased from W7 to W14, reflecting ongoing adaptation to the post-weaning environment. The enrichment of fiber-degrading bacteria, including Prevotella, Anaerovibrio, Treponema, and Muribaculaceae highlighted the establishment of a solid feed-adapted microbiota capable of degrading complex polysaccharides.
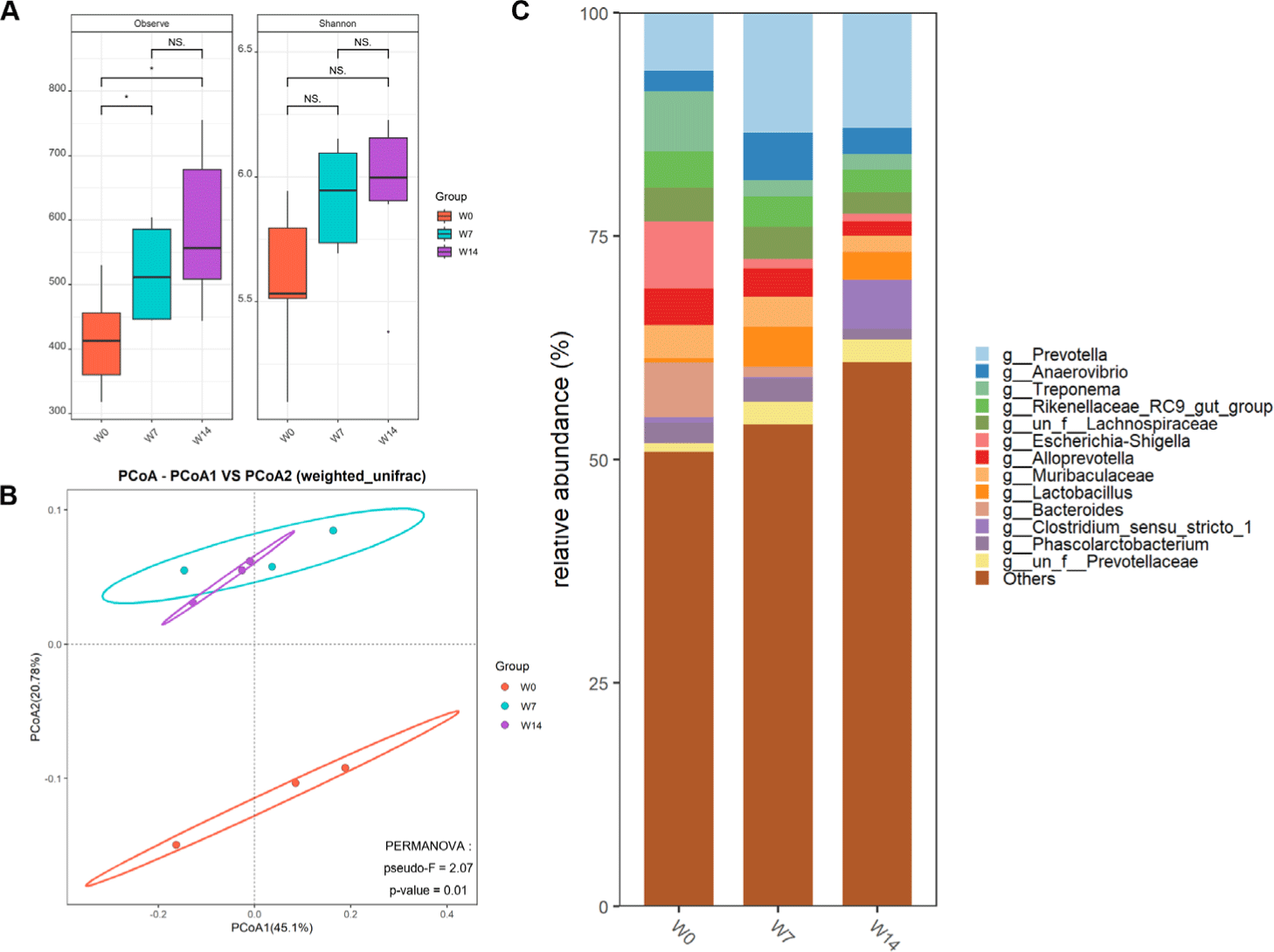
After weaning, potentially pathogenic bacteria, such as Campylobacter and Shigella, associated with post-weaning diarrhea decreased in relative abundance. These changes reflect adaptation to the post-weaning environment. Given the stable adaptive microbiome at W14, this time point was selected for subsequent analyses of host-microbiome interactions during weaning.
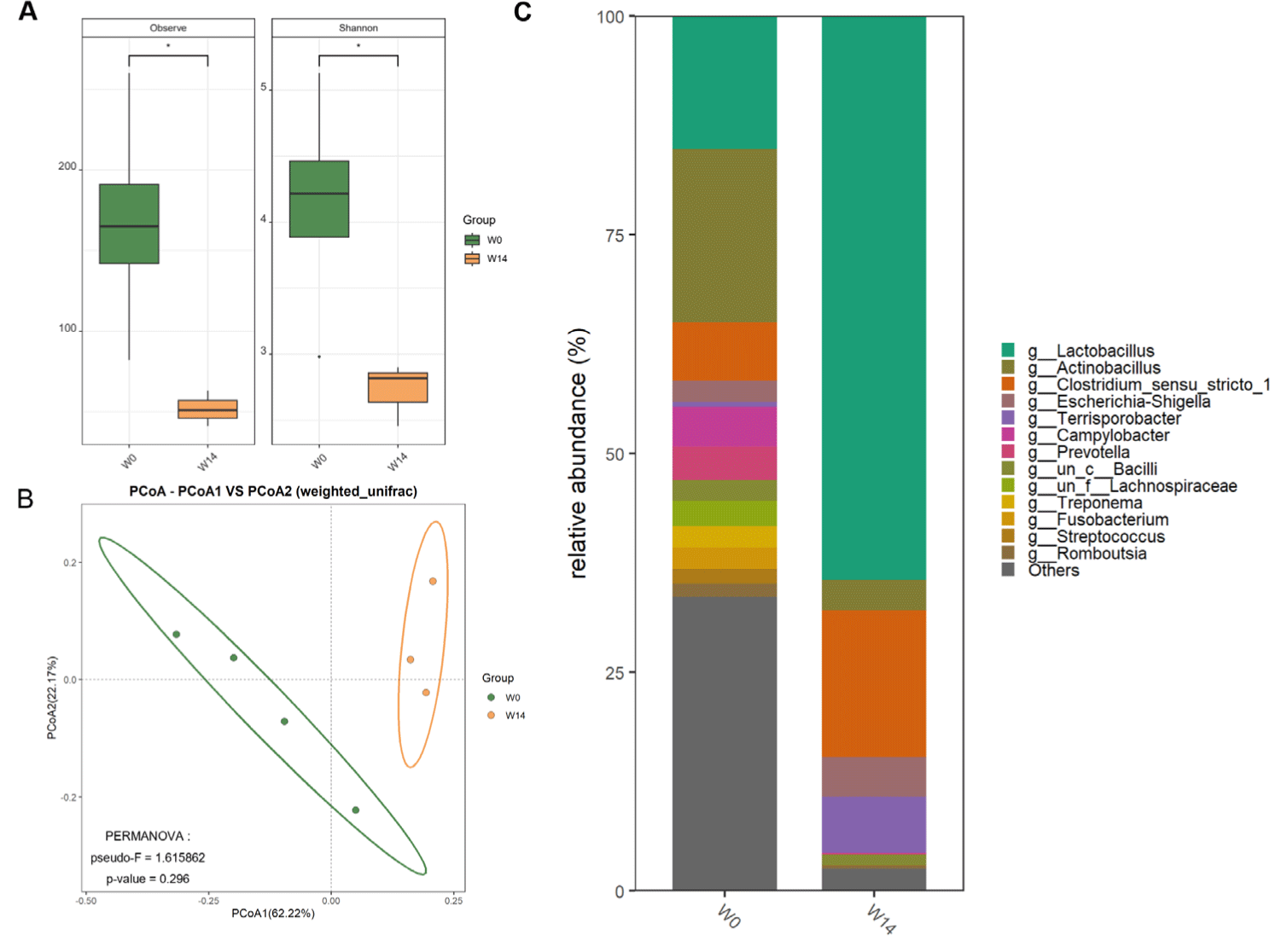
Analysis of the ileal microbiome represented 631,201 high-quality reads, with rarefaction curves indicating sufficient sequencing depth. ASVs (n = 1,218) were identified between W0 and W14. Alpha diversity analysis represented significantly higher species richness and Shannon diversity indices in W0 than in W14 (Fig. 4A). Beta diversity analysis using weighted UniFrac distances revealed distinct clustering patterns between W0 and W14, indicating substantial shifts in community composition after weaning (Fig. 4B). Genus-level taxonomic classification revealed substantial compositional shifts (Fig. 4C). Lactobacillus was dominant in W14, with higher relative abundances of Clostridium and Terrisporobacter compared to that in W0.
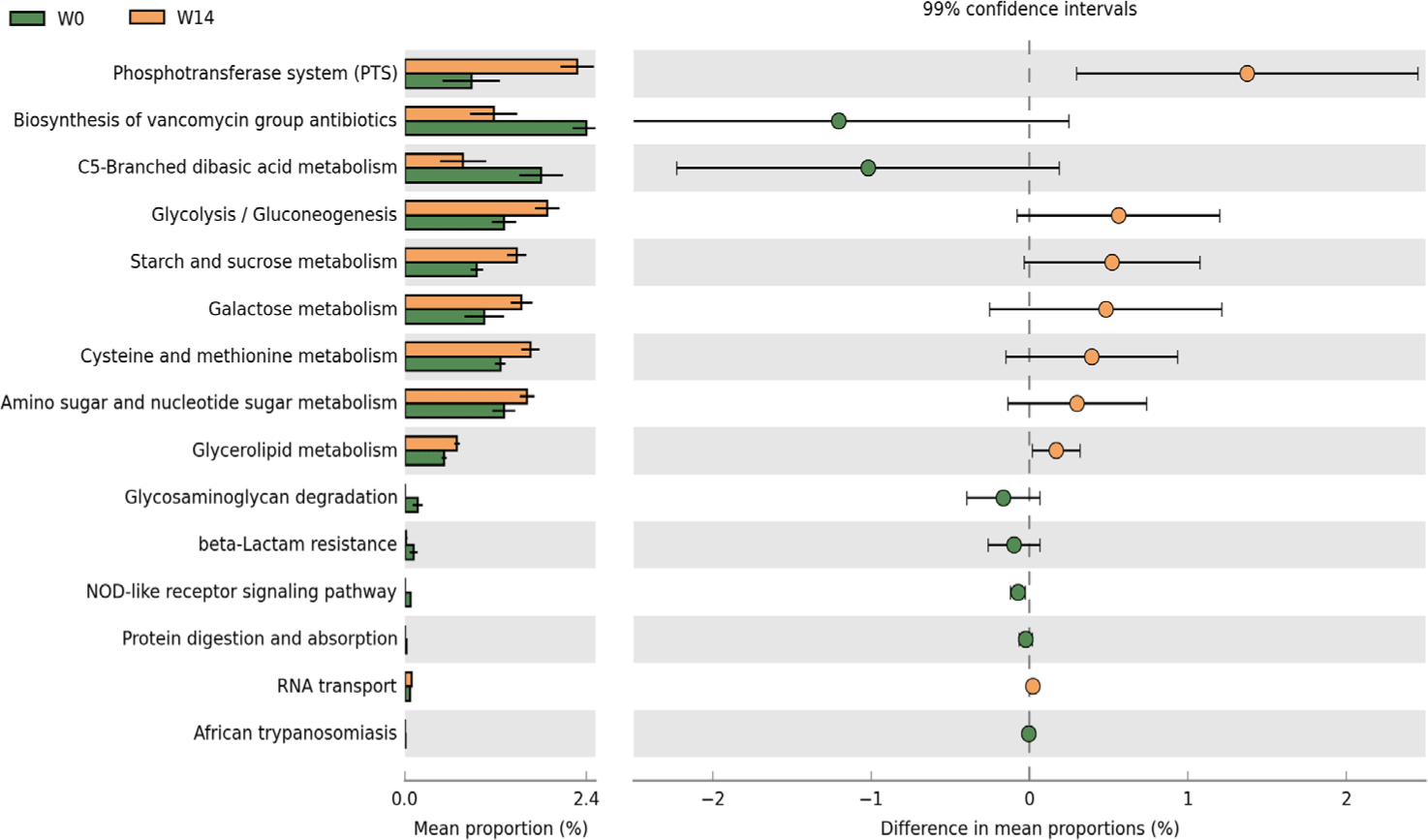
PICRUSt2 analysis predicted distinct functional ileal microbiota profiles at W0 and W14 (Fig. 5). The W14 microbiome was significantly enriched in multiple metabolic pathways, especially carbohydrate metabolism. Glycolysis/gluconeogenesis for energy production, and pathways for starch, sucrose, and galactose metabolism. Amino acid metabolism, especially cysteine and methionine pathways, was upregulated. Enrichment in glycerolipid and amino sugar/nucleotide sugar metabolism reflects the development of diverse metabolic capabilities. Conversely, the W0 microbiome was enriched in defensive and stress-response pathways, including vancomycin antibiotics biosynthesis for competitive survival, C5-branched dibasic acid metabolism for energy, and various defense mechanisms such as glycosaminoglycan degradation, beta-lactam resistance, and NOD-like receptor signaling. The functional shifts demonstrate a clear transition from a defense-responsive microbiome at weaning to a metabolically specialized community adapted for efficient nutrient utilization at 14 days post-weaning.
To predict potential metabolic relationships between microbial communities and metabolites using MIMOSA2 integration analysis was performed. CMP scores were calculated by integrating microbial abundance data with genome-scale metabolic models, which quantifies the community’s metabolic capabilities through both production and utilization of specific metabolites (Supplementary Files 3 and 4). The analysis revealed eight metabolites significantly influenced by microbial community changes (Fig. 6). L-serine, L-cysteine, glycine, uracil, linoleate, L-proline, 4-aminobutanoate, and ethanolamine showed significant associations with specific taxa (p < 0.1). For each metabolite, scatter plots demonstrate the relationship between CMP scores and metabolite measurements, with trend lines indicating the model fit. Bar plots reveal the taxonomic contributions to metabolite variance, where positive values indicate production potential and negative values suggest utilization. The results of MIMOSA2 analysis support mechanistically informed, hypothesis-generating associations rather than definitive cause and effect relationships.

Lactobacillus species emerged as major contributors to L-serine, L-cysteine, and L-proline metabolism, while Clostridiaceae significantly influenced glycine and uracil utilization. These relationships were identified through analysis of specific metabolic genes and reactions associated with each taxa-metabolite pair. Linoleate metabolism was predominantly influenced by a single taxonomic group, highlighting the specialized nature of certain metabolic processes in the ileal microbiome.
Morphological analysis of the ileal tissue sections showed changes in villus architecture between W0 and W14 (Table 1). Villus height was numerically higher at W14 than at W0 (227.14 µm vs 273.55 µm, p = 0.057). Crypt depth increased at W14 (184.83 µm vs 248.80 µm, p < 0.001), while the villus height to crypt depth ratio did not differ between time points (p = 0.789). Villus width decreased (119.28 µm vs 82.94 µm, p = 0.005), and villus area increased (16,717.82 µm² vs 21,910.66 µm², p = 0.012). Goblet cell counts were numerically higher at W14 (p = 0.055).
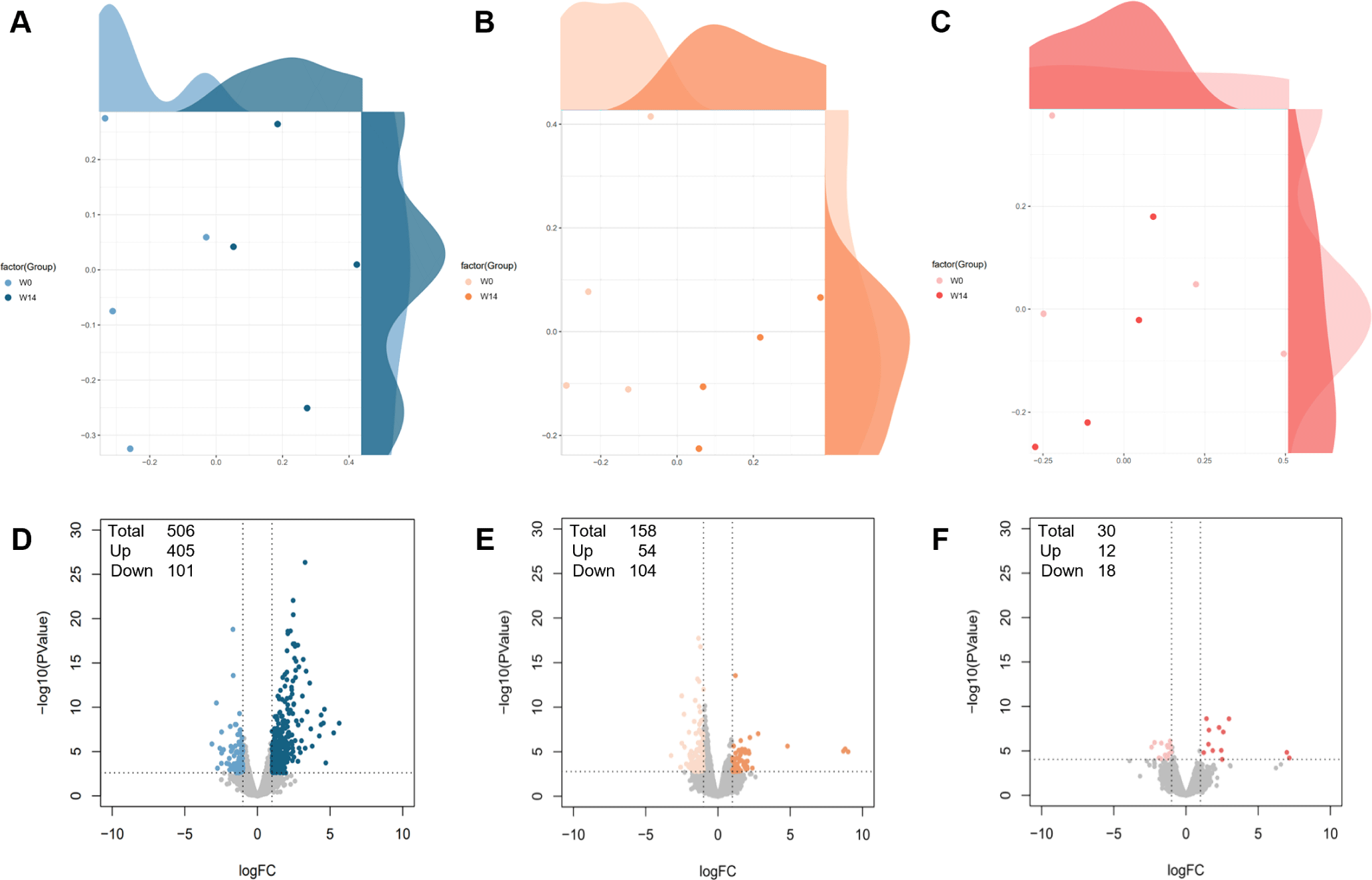
Transcriptional profiling was performed to analyze differential gene expressions between W0) and W14 in the ileum, thymus, and MLN. RNA sequencing yielded 20,700,935 raw reads (44.75% GC content), trimmed to 20,218,078 reads (44.38% GC content) (Table 2). Alignment analysis showed 86.48% uniquely aligned reads with a 94.91% overall alignment rate across 24 samples. MDS analysis revealed distinct expression patterns in the ileum and thymus, with minimal variation in the MLN between timepoints (Fig. 7A, 7B, and 7C). Differential expression analysis identified 506 DEGs (405 upregulated, 101 downregulated) in the ileum, 158 DEGs (54 upregulated, 104 downregulated) in the thymus, and 30 DEGs (12 upregulated, 18 downregulated) in the MLN (Fig. 7D, 7E, and 7F), defined by |Log2FC| ≥ 1 and FDR < 0.05.
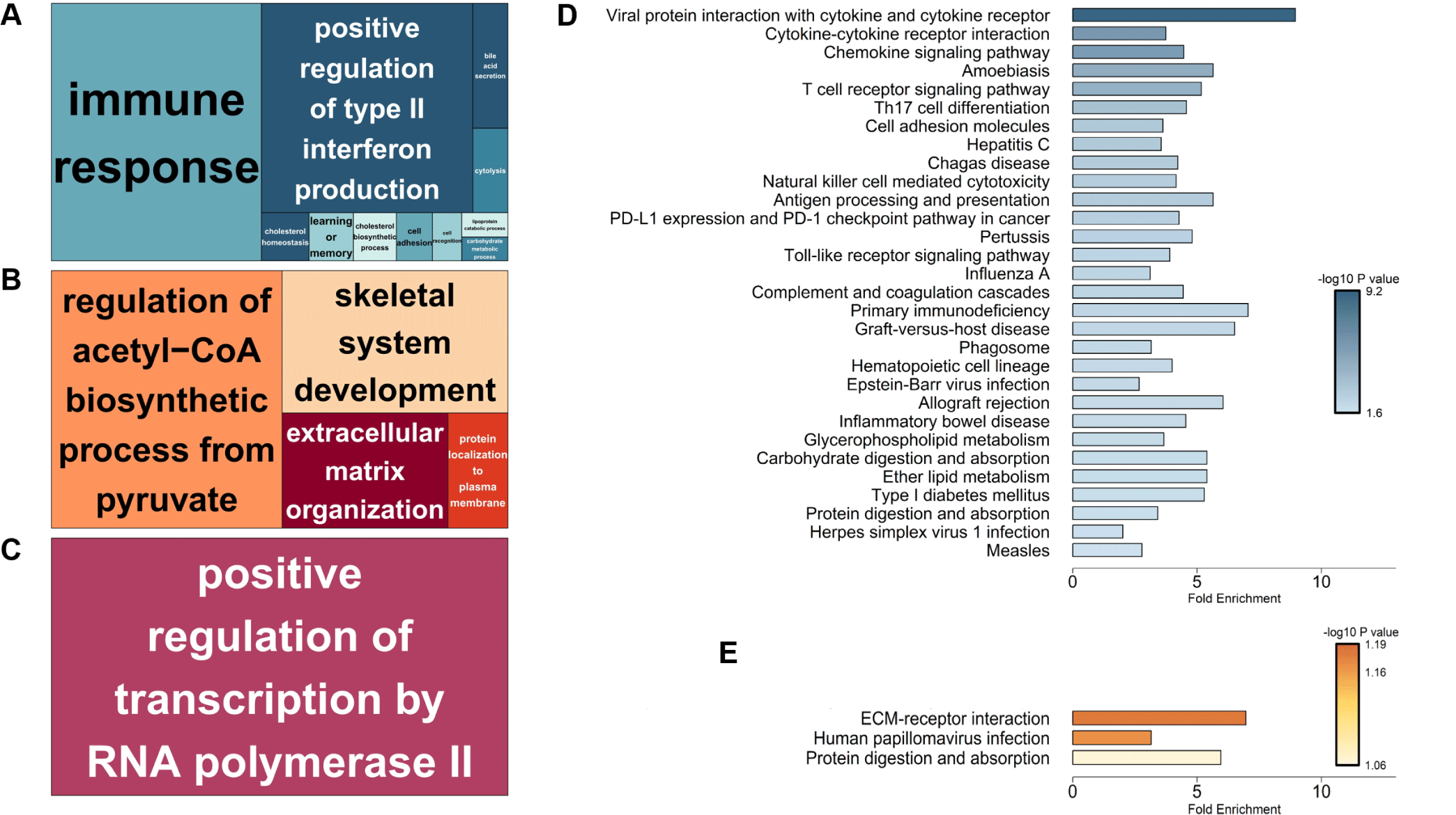
Functional enrichment analysis using the DAVID database revealed tissue-specific biological processes for DEGs. GO terms indicated predominant immune response and type II interferon production in the ileum, metabolic and skeletal system development in the thymus, and transcriptional regulation in the MLN (Fig. 8A, 8B, and 8C). KEGG pathway enrichment analysis revealed significant enrichment of ileum DEGs in immune-related pathways, including viral protein interactions with cytokine and cytokine receptors, chemokine signaling, and T cell receptor signaling. Barrier function-related pathways (cell adhesion molecules) and metabolism (glycerophospholipid metabolism, carbohydrate digestion, and absorption) were also significantly enriched (Fig. 8D). In the thymus, ECM-receptor interaction and protein digestion/absorption pathways were primarily enriched (Fig. 8E), while the MLN showed no significant KEGG pathways.
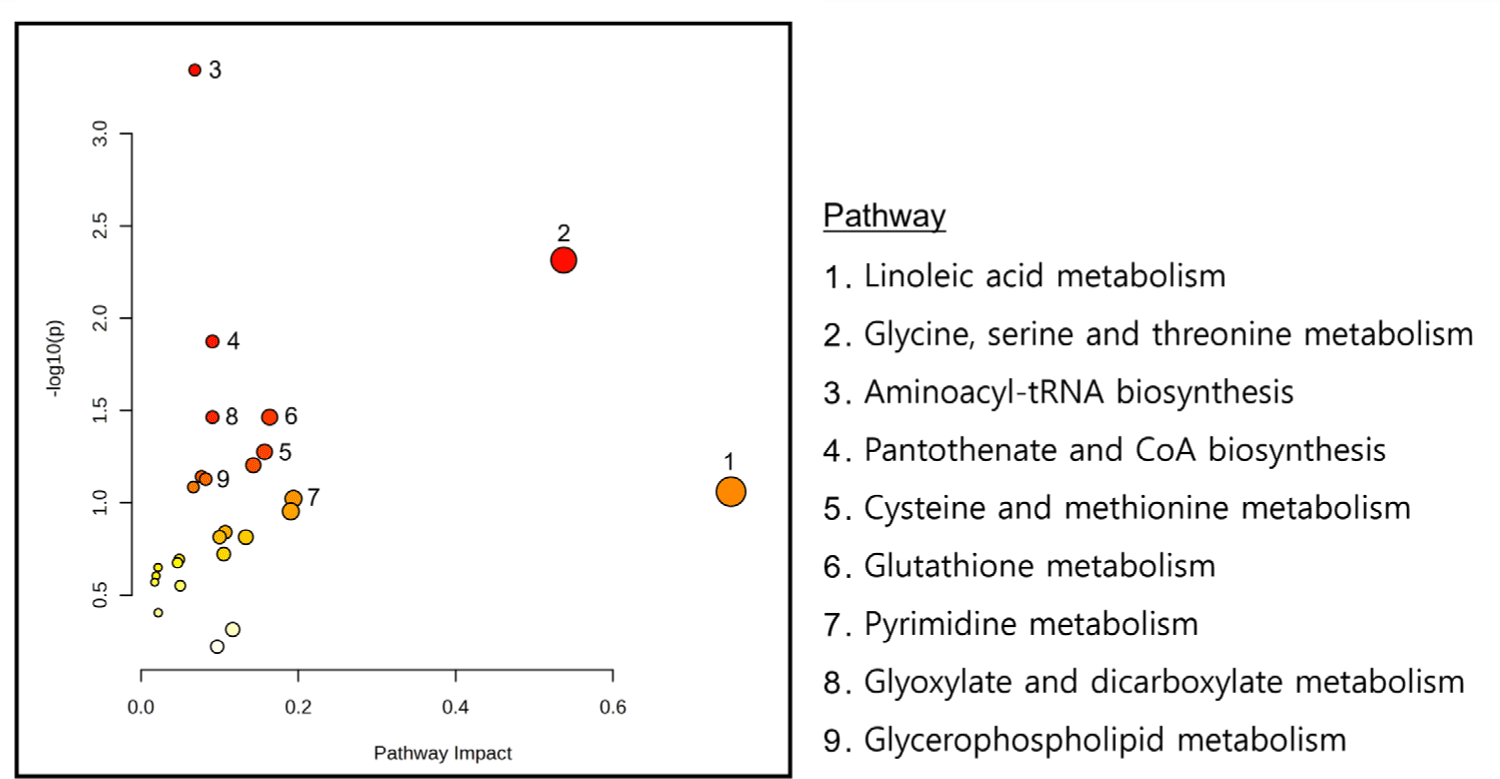
Joint pathway analysis was conducted in MetaboAnalyst 6.0 using ileal DEGs and putatively microbiome-governed metabolites identified by MIMOSA2 (Supplementary File 2). Commonly enriched pathways were identified using over-representation analysis with FDR adjustment, and pathway impact (Topology) was calculated using degree centrality. Nine pathways met the significance criteria in the joint pathway space (Fig. 9). Aminoacyl-tRNA biosynthesis showed the strongest enrichment signal, whereas linoleic acid metabolism showed the highest pathway impact score. Glycine, serine and threonine metabolism, cysteine and methionine metabolism, glutathione (GSH) metabolism, and glycerophospholipid metabolism were also identified among the enriched pathways.
The alluvial diagram revealed extensive interactions between ileal microbiota, microbial-derived metabolites, and host target pathways (Fig. 10). Lactobacillaceae predominantly contributed to proline, glycine, and serine metabolism, while Enterobacteriaceae, Turicibacteraceae, Veillonellaceae, and Pasteurellaceae contributed to ethanolamine, uracil, L-cysteine, and gamma-aminobutyric acid production.

Integration of microbe-derived metabolites into host metabolic pathways identified glycerophospholipid and GSH metabolism as key pathways for weaning adaptation. Glycerophospholipid metabolism, connected to proline and ethanolamine through LPCAT2, LPCAT3, and GPAT2 expression, was associated with intestinal epithelial remodeling and barrier function. GSH metabolism, linked to glycine and L-cysteine metabolism through GSTO1 expression, was involved in antioxidant defense and inflammatory response regulation in the intestinal mucosa. Additional pathways including pyrimidine, purine, arginine, and proline metabolism were also identified in the host-microbiome metabolic network.
DISCUSSION
Adaptation to weaning was consistent with the progressive establishment of a stable gut microbiome. Time-series analysis of fecal microbiota showed a significant shift in microbial composition, with diversity increasing from the weaning day to 14 days post-weaning (Fig. 3). The weaning transition was characterized by enrichment of fiber-degrading microorganisms, including Prevotella, Treponema, and members of the Muribaculaceae and Lachnospiraceae families [37]. A reduction in potentially pathogenic bacteria, including Campylobacter and Shigella, was consistent with development of a more balanced gut environment [38].
Ileal microbiome analysis at W14 indicated specialized microbial communities consistent with nutrient utilization during the dietary transition (Fig. 4). Dominance of Lactobacillus in the ileum and increased presence of fiber-degrading bacteria were consistent with adaptation to solid feed digestion [39]. The microbial shift was consistent with a transition from a milk-oriented community to a solid feed-adapted community [40].
Predicted functional profiling of ileal microbiota indicated a shift from stress-response features on the weaning day to increased metabolic capacities at 14 days post-weaning (Fig. 5). Enrichment of carbohydrate metabolism pathways, including the phosphotransferase system and sugar utilization modules, suggested functional adjustment for nutrient processing [41]. The predicted functional profile was consistent with coordinated changes in microbial composition and functional specialization during the weaning transition [42]. The 14-day post-weaning period may represent a time window for establishing a stable, functionally adapted gut microbiome, and the predicted functional changes were consistent with adaptation to the post-weaning environment [43].
Transcriptional profiling revealed distinct adaptation patterns across different tissues during the weaning transition, with the ileum exhibiting the most extensive molecular changes. The identification of 506 DEGs in the ileum, compared to that of 158 in the thymus and 30 in the MLN, highlights the ileum as a key site of adaptation during weaning (Fig. 7D, 7E, and 7F). This pronounced molecular response in the ileum reflects its critical role as the first point of contact with dietary changes and environmental challenges [44].
The significant enrichment of viral protein interaction with cytokine and cytokine receptor pathways likely reflects the heightened vulnerability of piglets during the weaning transition [45]. During this period, piglets encounter multiple challenges, including maternal antibody depletion, exposure to new environmental antigens, and dietary stress, all of which can compromise immune defenses [46]. The strong upregulation of interferon-induced chemokines (CXCL9, CXCL10, CXCL11) and their receptor CXCR3 establishes key antiviral defense mechanisms (Fig. 11) [47]. Enhanced antiviral response is crucial, as weaning stress increases susceptibility to viral infections [48].
Upregulation of inflammatory chemokines CCL11, CCL5, and CCL4, along with their receptors CCR2, CCR5, and CCR9, suggests the formation of a comprehensive immune surveillance system [49]. Immune network development appears to be an adaptive response to the increased risk of pathogen exposure during weaning while also promoting the establishment of immune tolerance to new dietary antigens [50]. The increased expression of IL18RAP and IL6ST reflects a balanced immune response, demonstrating the adaptation of the ileum to sustain immune homeostasis amid various environmental challenges [51]. The adaptation is crucial for defending against potential pathogens while preventing excessive inflammatory responses that could compromise gut barrier integrity [52].
Furthermore, the significant upregulation of components in the T cell receptor signaling pathway indicates the activation of adaptive immune responses in the ileum [53]. IFNG showed the strongest upregulation, and increased expression of CD3 complex components (CD3E, CD3D) and CD8A was consistent with T cell activation and differentiation (Fig. 11B). The upregulation of T cell co-stimulatory molecule CD28 and its regulatory counterpart CTLA4 suggests the establishment of balanced T cell responses [54]. This is particularly crucial during the weaning transition, as it supports proper immune responses and prevents excessive inflammation [48]. Expression of signaling molecules including ITK, FYN, and GRAP2 further supported involvement of T cell signaling pathways [55]. Taken together with the chemokine responses, the transcriptional profile was consistent with coordinated immune activation in the ileum during weaning [56].
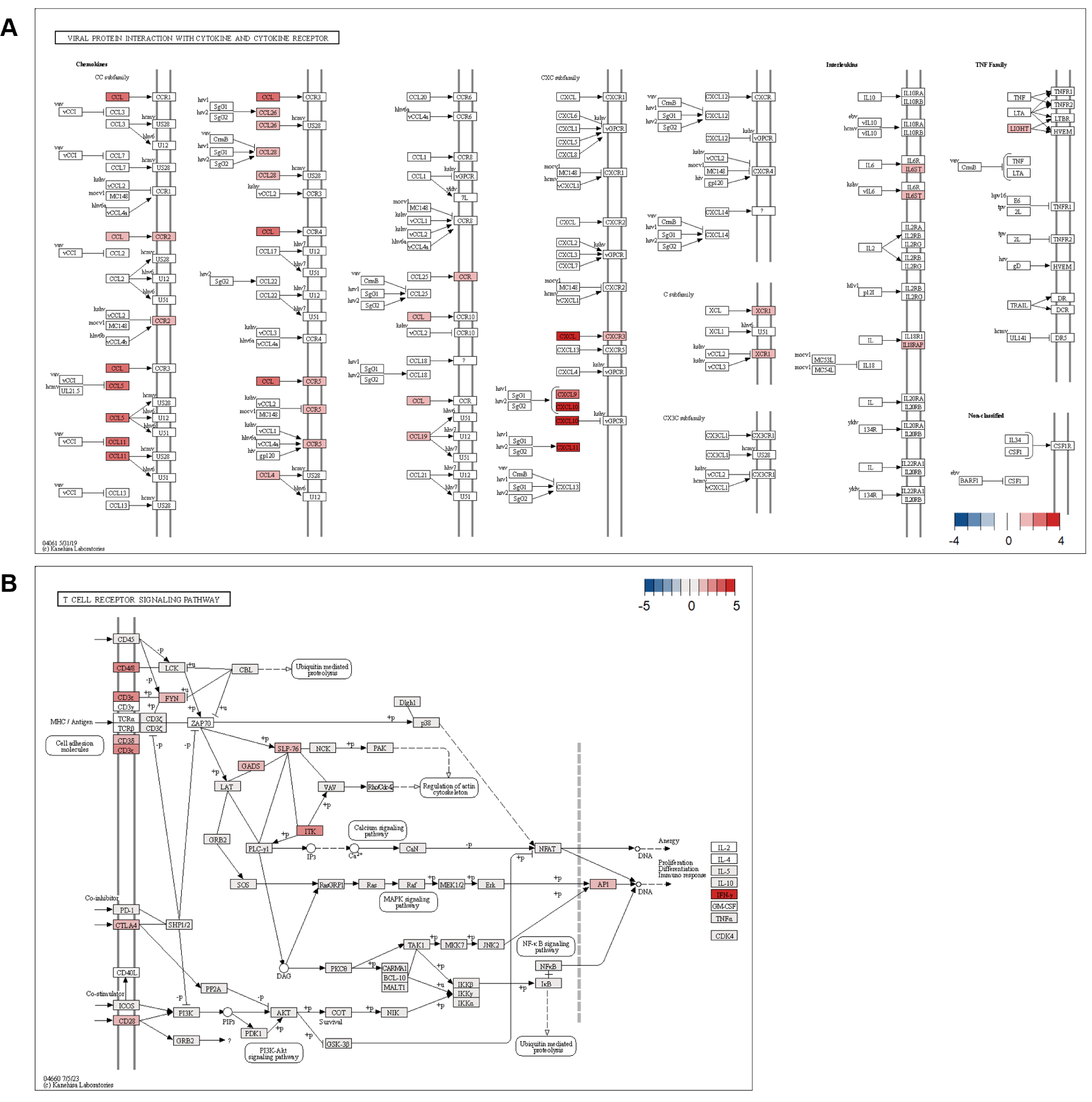
The simultaneous activation of these pathways suggests the establishment of both immediate and long-term adaptive immune responses, crucial for maintaining intestinal homeostasis during the challenging weaning transition period [57]. The thymus showed changes associated with piglet growth, including ECM-receptor interaction and skeletal system development. The MLN showed enrichment of positive regulation of RNA polymerase II transcription. Given the role of MLNs [58], immune cell activity may have increased in response to the altered intestinal environment after weaning. The tissue-specific patterns suggested that gut-localized immunity predominated over systemic responses during adaptation to weaning.
Host–microbiome features in the ileum during the weaning transition were associated with metabolic patterns, with glycerophospholipid and GSH metabolism highlighted as pathways potentially linked to barrier-related remodeling and antioxidant responses [59,60]. Integration analysis linked metabolites and gene expression features to these pathways, supporting a model-consistent interpretation of multi-omics integration.
The glycerophospholipid metabolism pathway showed regulation across multiple lipid-processing enzymes (Fig. 12A) [61]. GPAT3 (glycerol-3-phosphate acyltransferase 3) catalyzes an initial step by converting glycerol-3-phosphate to 1-acyl-sn-glycerol-3P [62]. The GPAT3 step is followed by CDS1 (CDP–diacylglycerol synthase), which mediates formation of CDP–diacylglycerol, a key intermediate in phospholipid biosynthesis [63]. LPCAT2/3 (lysophosphatidylcholine acyltransferases 2/3) facilitates phospholipid remodeling through acyl chain modifications [64]. PLB1 (phospholipase B1) and PLA2G12B (phospholipase A2 group 12B) may contribute to phospholipid homeostasis through hydrolysis and membrane remodeling [65]. Decreased expression of ETNK2 (ethanolamine kinase 2) was consistent with reduced phosphorylation of ethanolamine in the Kennedy pathway, which may relate to utilization of microbial-associated metabolites [66]. Taxon contribution outputs suggested differential predicted contributions to ethanolamine levels, with Lactobacillaceae associated with higher predicted contributions and Clostridiaceae associated with lower predicted contributions. Linoleic acid metabolism may integrate with glycerophospholipid pathways and may relate to membrane adaptation processes [67]. Enterobacteriaceae were linked to linoleate measurements in the intestinal environment, and upregulation of host phospholipases (PLA2G12B and PLB1) was consistent with glycerophospholipid metabolism potentially linked to barrier function [68,69]. The combined microbial and host patterns may represent aspects of adaptation during the weaning transition.
GSH metabolism showed an integrated pattern that aligned with glycerophospholipid metabolism through precursor availability and enzymatic activity (Fig. 12B). Taxon-level contribution outputs suggested associations between Clostridiaceae and L-cysteine production and between Lactobacillaceae and glycine utilization, which may relate to GSH synthesis through gamma-glutamyl cycle activity [70]. Host responses included upregulation of enzymes involved in antioxidant defense and xenobiotic detoxification, including GPX and GSTO1. GPX catalyzes conversion of reduced GSH to oxidized glutathione (GSSG) [71]. GPX activity neutralizes hydrogen peroxide (H2O2) and contributes to antioxidant defense [72]. The GPX-associated response may represent a component of weaning-associated stress responses, which have been linked to increased reactive oxygen species in intestinal epithelial cells [73]. GSTO1-mediated GSH conjugation may contribute to detoxification during dietary transition [74].
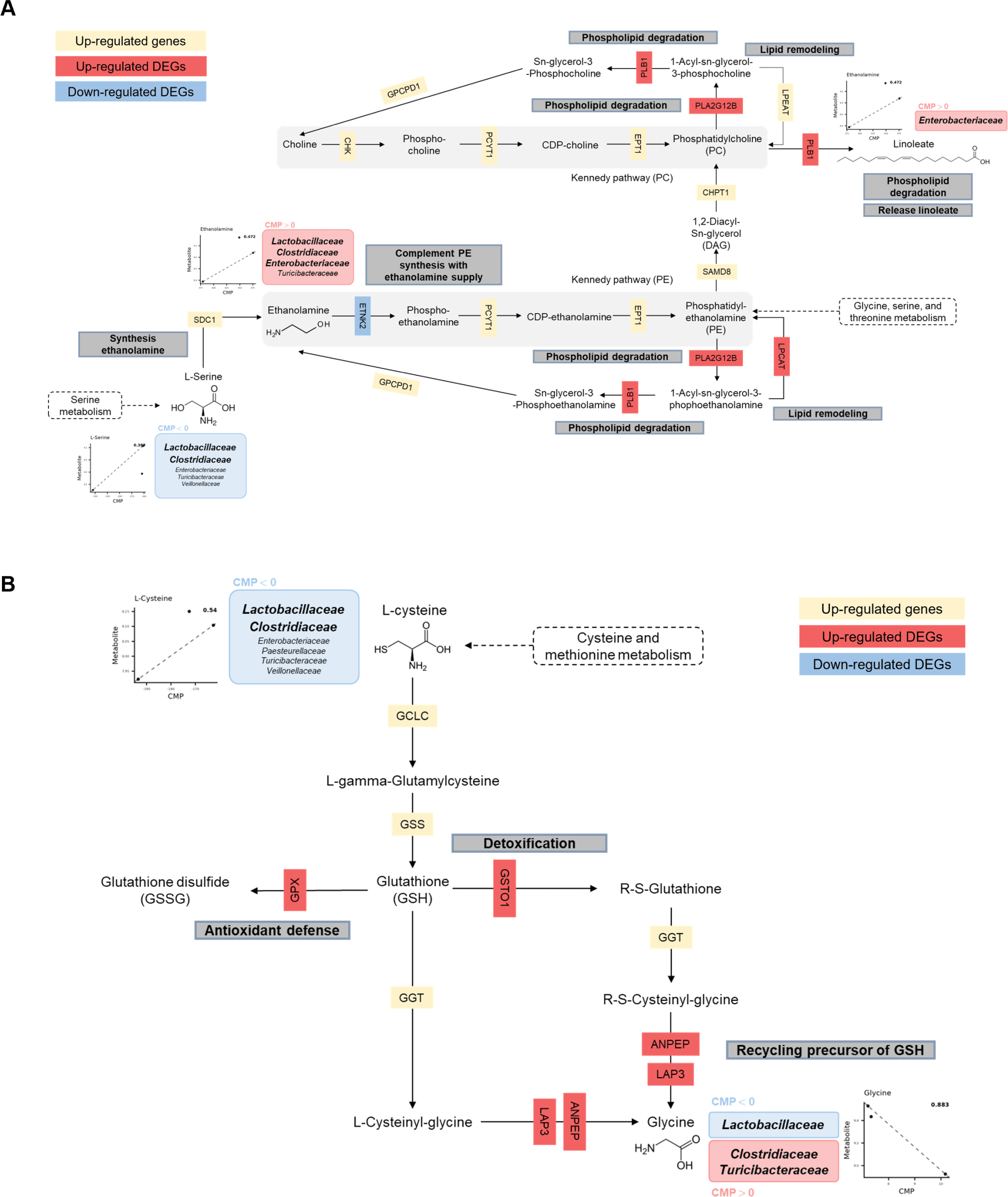
Regulation of GSH synthesis and utilization may contribute to redox homeostasis during weaning. The gamma-glutamyl cycle, supported by microbial-associated amino acids, may help maintain GSH availability for antioxidant activity, which is relevant to intestinal redox homeostasis and barrier function during weaning stress [75]. Upregulation of GSH metabolism-related genes, together with microbiome-associated precursor availability, was consistent with an antioxidant defense response by 14 days post-weaning.
Although pathways including pyrimidine metabolism, purine metabolism, and cAMP signaling were identified, glycerophospholipid and GSH metabolism showed more extensive integration between microbial metabolites and host responses. Enzyme and metabolite patterns in these pathways suggested potential roles during the post-weaning period. Glycerophospholipid metabolism may relate to barrier-associated lipid remodeling, and GSH metabolism may contribute to protection against oxidative stress and foreign substances. Together, the two pathways may contribute to intestinal resilience during the weaning transition.
Morphological analysis of the ileum showed villus-crypt changes during weaning that were consistent with intestinal adaptation. Villus height showed an increasing trend at W14, which may be consistent with increased absorptive surface area, and the change aligned with microbiome and metabolic profiles [76]. An increase in villus height may support the transition to solid feed digestion by increasing surface area available for nutrient absorption [77]. Crypt depth increased, which may be consistent with increased epithelial turnover and regenerative activity [78]. Increased crypt depth may contribute to epithelial renewal, which is relevant to barrier integrity during weaning-associated stress [79]. The villus height to crypt depth ratio (V/C ratio) showed a slight decrease, which was consistent with proportional remodeling during increased turnover [80]. The villus-crypt patterns were consistent with molecular and microbial features observed in the ileum. The villus-crypt changes were aligned with enrichment of carbohydrate metabolism pathways in the ileal microbiota and with upregulation of nutrient transport and metabolic genes in the host transcriptome [81]. Increased absorptive surface area may support establishment of fiber-degrading bacterial communities and may influence nutrient extraction from solid feed [82].
The villus-crypt patterns were consistent with barrier-related interpretations based on glycerophospholipid and GSH metabolism. Increased crypt depth provides a cellular basis for epithelial renewal [83], which is relevant to barrier maintenance under oxidative and inflammatory challenges during weaning. The morphological evidence supported an integrated interpretation of physical and molecular features during development of a functional gut barrier during the weaning period.
The results may inform commercial swine production and veterinary medicine. Identification of time-series metabolic features during weaning may inform nutritional targets for intervention. Glycerophospholipid and GSH metabolism may provide candidate pathways for developing feed additives or supplements that support barrier-related and antioxidant responses during the transition. Microbial shifts associated with intestinal adaptation may also inform probiotic development targeted to weaning piglets. The time-series patterns of microbial colonization and host responses observed in the study may help refine timing of dietary interventions and management practices, which may reduce weaning-associated production losses and may improve animal welfare. Future studies should evaluate translation of the molecular patterns into nutritional and management strategies for piglet health during the weaning transition.
CONCLUSION
We characterized host–microbiome features during the weaning transition in piglets by integrating time-series fecal microbiome profiling (W0, W7, W14) with paired ileal microbiome, metabolome, and tissue transcriptome measurements (W0, W14). Fecal microbiota showed a compositional shift after weaning, with increased diversity by W14 and enrichment of fiber-degrading taxa including Prevotella, Treponema, Muribaculaceae, and Lachnospiraceae, together with reduced relative abundance of Campylobacter and Shigella. In the ileum, community structure differed between W0 and W14, with lower alpha diversity at W14 and predominance of Lactobacillus, accompanied by predicted functional enrichment of carbohydrate and amino acid metabolism at W14. Ileal transcriptomic responses were most pronounced, with 506 DEGs and enrichment of immune-related pathways including viral protein interaction with cytokine and cytokine receptor and T cell receptor signaling, whereas thymus and MLNs showed fewer DEGs. Ileal morphology showed villus–crypt remodeling from W0 to W14, including increased crypt depth (p < 0.001) and a trend toward higher villus height at W14 (p = 0.057). MIMOSA2 identified putatively microbiome-governed metabolites from CMP–metabolite associations (p < 0.1) and taxon contribution outputs, and joint pathway analysis highlighted glycerophospholipid and GSH metabolism among pathways linking ileal DEGs with microbiota-derived metabolites.
These results provide a hypothesis-generating framework in which weaning-associated microbiome restructuring is accompanied by coordinated ileal immune transcriptional programs and metabolic patterns consistent with membrane remodeling and redox homeostasis. The integrated signals may inform future nutritional or microbiome-directed strategies to support piglets during the weaning transition.