













INTRODUCTION
Copper (Cu), which is one of the essential trace elements that plays an crucial role in various ways in physiological processes. It is known to perform oxidation-reduction reactions, transport of oxygen and electrons, and protection against oxidative stress [1,2]. Copper in feed is reported to improve body weight gain and feed conversion ratio and reduce the frequency of diarrhea [3]. Copper can be included at growth-promoting levels (i.e, 75 to 250 mg/kg diet) in diets for weaning and growing pigs to reduce post-weaning diarrhea and improve growth performance [2]. Not only it improves growth performance, but also has an effect on intestinal microbes by enhancing host bacterial resistance [4].
Copper may be provided in sulfate, oxide, or chloride forms, as chelated Cu, or as Cu hydroxychloride in order to induce a faster growth and a better resistance to various diseases, which, to a great extent, correlated with the improved metal bio-availability [5,6]. Copper sulfate (CuSO4) is the most common form of inorganic copper suppplement in animal feeds due to its availability, and its relatively low cost compared with other sources of copper [7], which enhances growth performance and gut health [8,9]. On the other hand, organic copper such as copper amino acids, which improve apparent total tract digestibility and retention of copper in pigs by preventing formation of insoluble complexes along the gastrointestinal tract [10]. In addition, copper hydroxylchloride, which is as effective as CuSO4 in enhancing growth rate and feed efficiency in pigs [11], including low toxicity compared to CuSO4 [12]. However, supplementation of copper may result in fecal copper excretion [13]. The increased copper excretion might accumulate in soil and water, resulting in reduction of crop yield and potential toxicity for livestock [13]. Therefore, this study evaluates the effects of different types of copper supplementation on the intestinal microbiome of pigs.
MATERIALS AND METHODS
The experimental protocol for this study was reviewed and approved by the Rural Development Administration, Jeonju, Republic of Korea (approval Number NIAS-2020-1849). A total of 40 growing pigs (Duroc × Landrace × Yorkshire; Male, 73 ± 1 day of old) were alloted to a completely randomized block design. The pigs (average initial body weight of 30.22 ± 1.92 kg) were individually placed in 1.1 × 3.5 m2 stainless steel metabolism cages in an environmentally controlled room (22 ± 1°C). There were 2 pigs per treatments in a cage and 4 replicate cages per treatments. The dietary treatments consisted of CON (No additional added copper sulfate in diet), CuSO4 (positive control; CON + 100 ppm/kg copper sulfate), Cu-glycine complex (CuGly; CON + 100 ppm/kg of copper with glycine), and Cu-amino acid complex (CuAA; CON + 100 ppm/kg of copper with amino acids; Proline, Glutamic acid, Serine, Glycine, Aspartic acid, Arginine, Valine, Leucine, Threonine, Alanine, Lysine, Methionine, Isoleucine, Tryosine, Phenylalanine, Histidine, and Cystine), and Cu-hydroxy(4methylthio)butanoate chelate complex (CuHMB; CON + 100 ppm/kg of copper chelate). All diets were formulated to meet or exceed the National Research Council (NRC) (Table 1). Throughout the entire experimental period, pigs had ad libitum access to feed and water.
1) Supplied per kg of diet: Vit A, 5,000,000 IU; Vit D3, 1,000,000 IU; Vit E, 1,000 mg; Vit B1, 150 mg; Vit B2, 300 mg; Vit B12, 1,500 mg; niacin amide, 1,500 mg; DL-calcium pantothenate, 1,000 mg; folic acid, 200 mg; Vit H, 10 mg; choline chloride, 2,000 mg; Mn, 3,800 mg; Zn, 1,500 mg; Fe, 4,000 mg; Cu, 500 mg; I, 250 mg; Co, 100 mg; Mg, 200 mg.
A total of 80 fecal samples were collected directly from the rectum of the 40 pigs before (week 0) and after dietary supplementation (week 4) (8 pigs per group: CON, CuSO4, CuGly, CuAA, and CuHMB). Total DNA from the feces was extracted from 200 mg of feces per sample using QIAamp Fast DNA Stool Mini Kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. The concentrations of DNA were measured using a Colibri Microvolume Spectrometer (Titertek Berthold, Pforzheim, Germany) and samples with OD260/280 ratios of 1.80–2.15 were processed further. The V3 to V4 hypervariable regions of the 16S rRNA gene were amplified using the universal primers 341F (5’-TCGTCGGCAGCGTCAGATGTGTATAAGA GACAGCCTACGGGNGGCWGCAG-3’) and 805R (5’-GTCTCGTGGGCTCGGAGATG TGTATAAGAGACAGGACTACHVGGGTATCTAATCC-3’). The 16S rRNA gene libray preparation was performed, and MiSeq sequencing of 16S rRNA gene amplicons was conducted using the Illumina MiSeq platform at Macrogen (Seoul, Korea). Briefly, random fragments of DNA samples followed by 5’ and 3’ adapter ligation was conducted to prepared the sequencing library. Using the Nextera XT Index Kit (Illumina, San Diego, CA, USA), dual indices and Illumina sequencing adapters were attached to the 16S rRNA gene amplicons. The concentration of final products was normalized and pooled, using PicoGreen (Turner BioSystems, Sunnyvale, CA, USA), then the verification of the size of libraries was conducted using the TapeStation DNA ScreenTape D1000 (Agilent Technologies, Santa Clara, CA, USA). The polymerase chain reaction (PCR) cycle conditions included a 3 min initial denaturation at 95°C, 8 amplification cycles (95°C for 30s, 55°C for 30s and 72°C for 30s) and a 5 min final elongation at 72°C.
Mothur software was used to remove low-quality sequences from the 16S rRNA gene sequences. Briefly, sequences that did not match the PCR primers were discarded from the demultiplexed sequence reads. To minimize the effects of random sequencing errors, sequences containing ambiguous base calls and sequences with a length of less than 100 bp were trimmed. Chimeric sequences were eliminated using the UCHIME algo-rithm implemented in Mothur. With an operational taxonomic unit (OTU) definition at an identity cutoff of 97%, de novo OTU clustering was conducted using the Quantitative Insights into Microbial Ecology (QIIME) software package (version 1.9.1) [14]. The naïve Bayesian RDP classifier and the Greengenes reference database were used for the taxonomic assignment of the sequences. Beta-diversity was measured using unweighted UniFrac distance metrics using QIIME, which considers community membership (pres-ence or absence of OTUs). Principal coordinate analysis (PCoA) plots were generated based on unweighted UniFrac distance metrics.
The gut microbiome parameters were calculated using the R package MicrobiomeAnalystR and GraphPad Prism version 7.00 for Windows (GraphPad Software, CA, USA). Orthogonal contrasts (CON vs. other treatments; CuSO4 vs. other treatments) were used to compare the possible relationships between the treatments. A two-way ANOVA was used to confirm the alpha-diversity and taxonomic analysis data. Differences among treatment means were determined using Tukey’s test. For a two-sided Welch’s t-test in the Satistical Analysis of Metagenomic Profiles (STAMP) software v2.1.3 was used to identify significant differences in the relative abundance of micrbial taxa among the five groups. Analysis of similarities (ANOSIM) was used to determine wheter the microbial compositions between the five groups were significantly different or not using QIIME and were based on the unweighted and weighted UniFrac distance metrics. Statistical significance was set a p < 0.05.
RESULTS
Total DNA was extracted from fecal samples of the pigs, and the extracted DNA was amplified and sequenced through PCR, using primers specific to the V3 to V4 hypervariable regions of the 16S rRNA genes. Sequencing of the 16S rRNA genes in fecal samples produced a total of 10,761,929 reads, which ranged from 108,083 to 163,252 reads per sample. The average quality score (Phred scores) across all the samples ranged from 32 to 36. Further data filtering was performed to remove low quality or uninformative data and to improve the downstream statistical analysis. It resulted in 7,325,950 combined reads from all the samples, with an average reads per sample of 91,574.
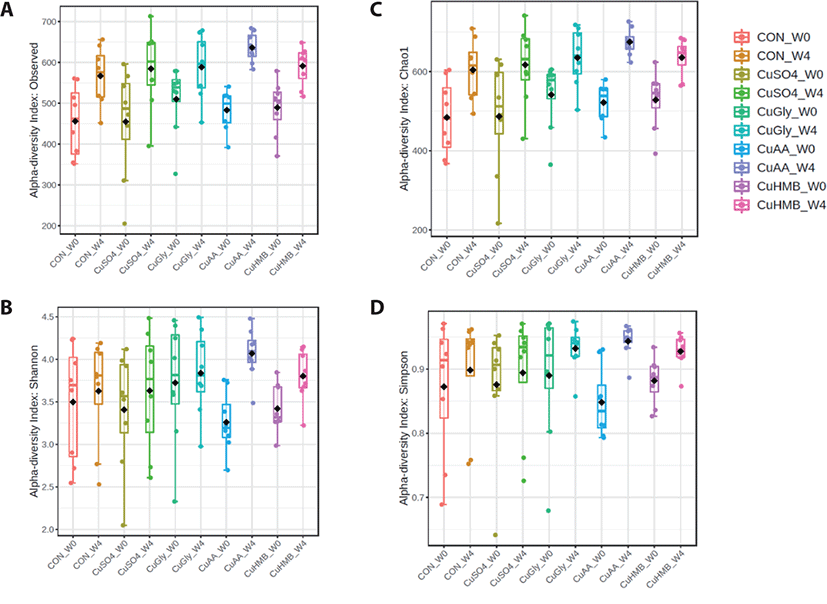
From the fecal samples, the diversity of the microbial coummunities of pigs was measured using: species richness (number of observed OTUs and Chao1) and species evenness (Shannon and Simpson) within a sample (Fig. 1). Among five groups at week 0, the number of observed OTUs ranged from 205–595, Chao1 as 220.75–628.06, Shannon and Simpson 2.05–4.45 to 0.64–0.97, respectively. CuGly group showed the highest number of observed OTUs, Chao1, Shannon, and Simpson indices with mean values of 511.12 ± 86.56, 548.91 ± 93.21, 3.72 ± 0.72, and 0.89 ± 0.10, respectively. At week 4, 442–703 ranging on the number of observed OTUs, 429.77–471.87 in Chao1, Shannon and Simpson 2.52–4.50 to 0.72–0.97, respectively. CuAA group showed the highest number of all indices with mean values of 639.75 ± 34.16, 675.32 ± 36.38, 4.07 ± 0.29, and 0.94 ± 0.02, respectively. It indicates that supplementation of CuAA group increased the species richness and diversity in the feces of pigs at week 4. However, no statistical differences were shown with Shannon and Simpson index at both week 0 and 4. Conversely, CuAA group at week 4 was observed significantly different with observed OTUs and Chao1 as compared to the CON group (p < 0.05).
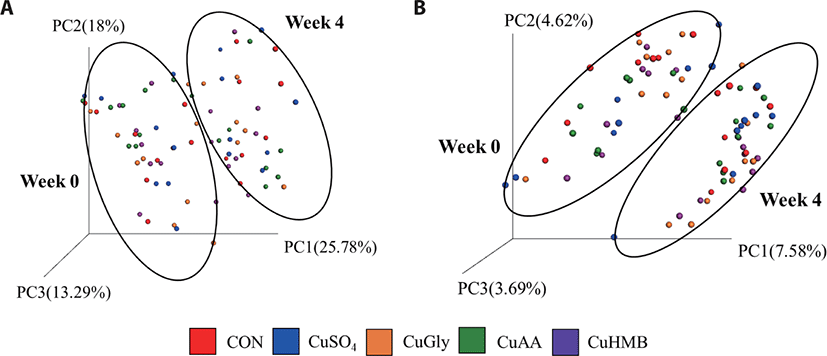
At week 0, the PCoA plot showed no significant differences of the microbial community among control, positive control and treatment groups as confirmed by ANOSIM, from results of both weighted and unweighted UniFrac distances, (p > 0.05, data not shown). Conversely, each group clustered significantly by weeks (week 0 and 4) which suggests that the microbial community structure shifts over time regardless of the treatment (Fig. 2).
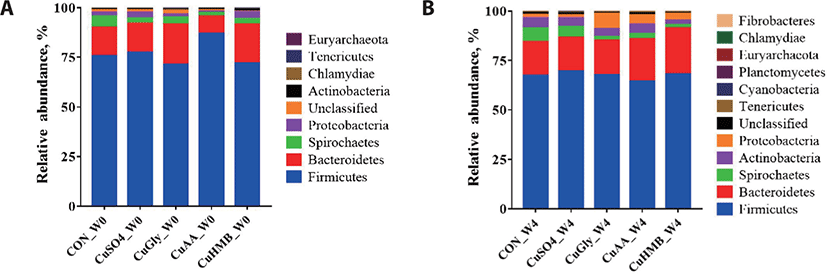
Microbial composition of swine gut with copper supplementation was determined. The relative abundance of different bacterial taxa at the phylum level among the five groups (CON, CuSO4, CuGly, CuAA, and CuHMB) between week 0 and week 4 (Fig. 3). The most common phyla in all the samples at both week 0 and week 4 were Firmicutes and Bacteriodetes. At week 0, Firmicutes and Bacteroidetes in each groups were 75.72%, 14.59% (CON), 77.50%, 14.53% (CuSO4), 71.35%, 20.31% (CuGly), 87.09%, 8.74% (CuAA), and 71.94%, 19.82% (CuHMB), respectively. In addition, the relative abundance of Proteobacteria on CON group were 2.02%, followed by CuSO4 (2.89%), CuGly (1.52%), CuAA (0.94%), and CuHMB (3.35%). At week 4, Firmicutes and Bacteroidetes were also the most predominant phyla in all the samples, CuHMB group having the highest combined relative abundance of 91.39% followed by CuSO4 (86.53%), CuAA (85.76%), CuGly (85.23%), and CON (84.46%). Moreover, the relative abundance of Proteobacteria, CuGly had the highest and CuSO4 had the lowest proportions among 5 groups. These findings were similar to previous studies in swine gut microbiota, indicating that phylum Firmicutes and Bacteroidetes are the major phyla, regardless of copper supplementation treatment.
The relative abundance of different bacterial taxa at the genus level among the five groups (CON, CuSO4, CuGly, CuAA, and CuHMB) between week 0 and week 4 (Fig. 4). The bacterial genera with counts lower than 5,000 were merged into “Others”. At week 0, the top most predominant classified genera in the fecal microbiota of pigs were Lactobacillus. Lactobacillus with the highest and lowest abundance detected in the CuAA (58.52%) and CuGly (49.98%) groups. Nevertheless, the relative abundance of Lactobacillus has decreased on week 4, since other genera increased such as Megasphaera, Prevotella, and SMB53 in all five groups.
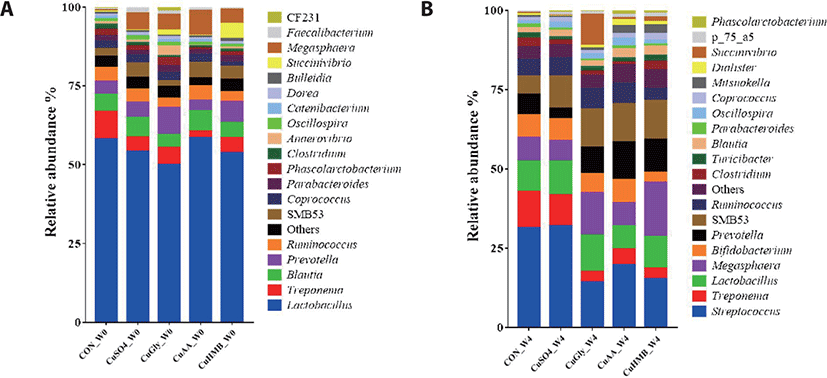
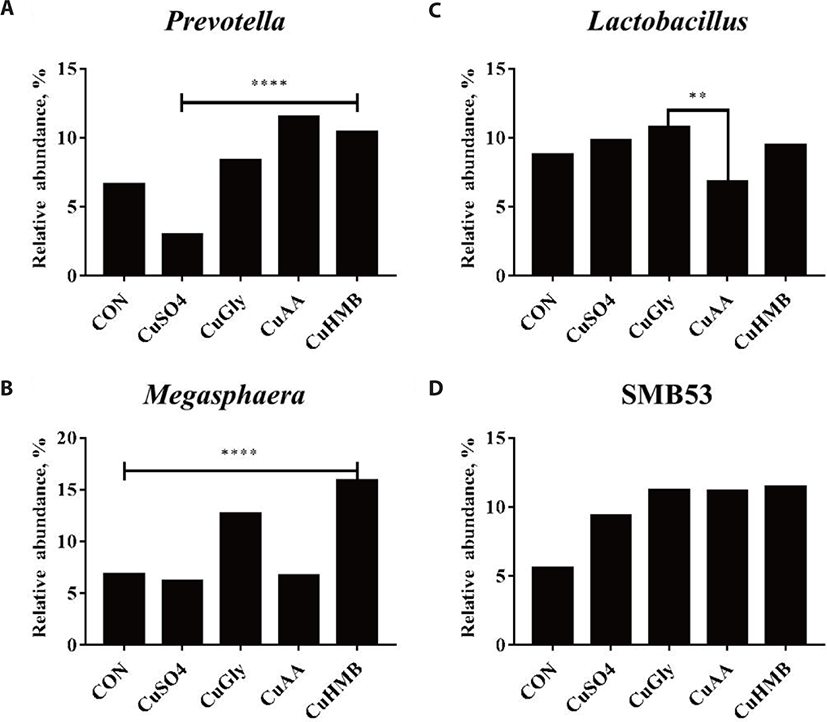
At the genus level, Lactobacillus was the most enriched genus in all fecal samples in week 0 (Fig. 4A), Streptococcus in week 4 (Fig. 4B) as followed. 4 genus were significantly different either CON or CuSO4 group in week 4; Prevotella, Lactobacillus, Megasphaera, and SMB53 (Fig. 5). Prevotella, its relative abundance increased significantly in the CuGly, CuAA, and CuHMB (8.35%, 11.51%, and 10.41%) groups compared to the CON and CuSO4 group (6.63% and 2.97%; Fig. 5A). The relative abundance of Lactobacillus significantly increased from an average of 10.75% and 9.46% in the CuGly and CuHMB group, whereas its abundance in the CuAA decreased (6.83%) compared to that in the CON group (8.77%; Fig. 5B). Megaspharea showed significantly high relative abundance in CuGly and CuHMB (12.71% and 15.85%) groups than other groups (CON; 6.85%, CuSO4; 6.18%, CuAA; 6.70%; Fig. 5C). For SMB53, all groups compared to CON group (5.56%) have increased significantly in realtive abundance (9.35%, 11.20%, 11.16%, and 11.44%, respectively; Fig. 5D).
DISCUSSION
Copper is an essential mineral in pigs with an essnetial cofactor for many cellular enzymes and many biological functions. It promotes bone formation, iron metabolism, enhances immunity and antioxidant capacity [4,15]. Copper supplementation (150–250 mg Cu/kg feed) in pig feeds has become popular as alternative to antibiotics due to the ban of antibiotic growth promoters [16,17]. Several studies reported that copper supplementation may not only regulate the activities of various enzymes such as digestive and antioxidant enzymes in pigs, but also may regulate the growth signalling pathways, feeding and fasting signalling pathways, and hence improve protein deposition and promote bone formation [18,19]. However, the overuse of CuSO4 results in an increase of copper content of pig slurry, resulting in copper in soils, which poses a high environmental risk [20]. Not only affecting the swine gut microbiota but also reduces the counts of Streptococci, ureolytic bacteria, and total anaerobes in the feces [4]. In order to reduce the usage of CuSO4, it is recommened to use the alternatives, which is known as organic copper. According to Association of American Feed Control Officials (AAFCO), organic minerals(copper) showed better absorbability, reduced excretion out of the body, stability in the gastrointestinal tract (GIT), and reduced toxicity compared to inorganic minerals such as CuSO4 [21]. Due to the high cost of organic copper compared to CuSO4, further studies are necessary to minimize use of copper in swine diets and prevent copper pollution in the environment.
Formal studies showed the mammalian GIT contains more than 500 bacterial species that would play important roles in the health and disease of the host [22]. The gut microbiota is being indicated to work importantly in the health and growth of animals, including the incidence decrease of infections, inflammations and other immune diseases [23,24]. As the attention to swine gut microbiome has increased, we performed this study to better understand microbial succession and fecal microbiome shifts in the growing pig by supplying different forms of copper for 4 weeks. The relative abundance of Prevotella significantly increased after supplement of CuAA and CuHMB groups compared to CuSO4 group. Generally, Prevotella is well known to produce enzymes, such as β-glucanase, mannase and xylase [25] which take part in degrading polysaccharides originated from plant cell walls into short-chain fatty acids (SCFAs), resulting in utilization as an energy source, including prevention of gut inflammation [26,27]. CuGly group has been identified as the highest relative abudance of Lactobacillus among all treatments of copper supplementation. Lactobacillus is the well-known bacterium to utilize carbohydrates with various genes encoded with a wide range of functional capacities related with carbohydrate transport and utilization [28]. CuGly and CuHMB groups had significantly higher relative abundance of Megaspharea, alike Prevotella, it can also degrade carbohydrates efficiently by converting into SCFAs, resulting in producing energy source for growth performance [29,30]. For SMB53, which belongs to the Clostridiaceae family, all copper supplementaion groups showed significantly higher relative abundance compared to CON and CuSO4 group. Previous studies indicated that SMB53 genes are related with immune system [31], and bile acid synthesis regulation [32,33]. Our results suggests that these significantly enriched genera may play a role in swine gut microbiota modulation which could contribute in energy production for enhancing growth performance, and regulating immune system, preventing from gut inflammation and other infections related to intestines.
Since there are some doubts mentioning about the structural integrity (and consequently metal bio-availability) of amino-acid chelates at low pH values of the first digestive tract, a new chelating ligand which is available to provide metal chelates sufficiently stable at low pH values is needed [34,35]. The supplementation of CuHMB, previous studies showed that it indicated more stable at low pH values than the corresponding chelates with amino-acids [36,37] with higher bioavailability as compared with the CuSO4 [38].
In this study, we evaluated the changes in the intestinal microbial communities induced by different forms of dietary copper supplements. It has been revealed that various types of organic copper supplements shift the intestinal microbiota in growing pigs and may provide insights into the development of feed additives to improve growth performance in swine.
CONCLUSION
The different forms of copper supplementation showed varying effects on the composition of the gut microbiota compared to the basal diet(CON) and copper sulfate(CuSO4) supplementation in growing pigs. CuGly and CuHMB could enhance beneficial microbiota that contribute to the degradation of polysaccharides and carbohydrates of plant cell walls into SCFAs, which can be converted to an energy source and prevent intestinal inflammation, resulting in improved growth performance.