













INTRODUCTION
Hanwoo cattle is the most dominant breed in Korea and has been genetically selected and improved as beef cattle with a high marbling content [1]. In the first quarter of 2022, the population of Hanwoo cattle was 3.34 million, which represented > 90% of all beef cattle raised in Korea. Hanwoo cattle are fed based on one of two feeding systems: separate feeding and total mixed ration (TMR) feeding. Since TMR feeding has advantages such as accuracy, convenience, and less feed selection, the use of the TMR feeding system has gradually increased and accounts for more than 20% of all Hanwoo farms in Korea [2].
Diverse microbiota is harbored in the gastrointestinal tract and contributes to nutrient digestion though fermentation. Therefore, gastrointestinal microbiota is important for the health of the host [3]. Cattle fecal microbiota is well known as one of the important factors affecting the health, productivity, and well-being of the host [3–5]. The 16S rRNA-gene based next generation sequencing analysis has been applied to investigate the fecal microbiota composition in cattle. Initially, pyrosequencing on the Roche GS FLX was used to evaluate the fecal microbiota composition in cattle fed different diets [6–9], but the number of sequence reads analyzed in these studies was limited. Recently, the Illumina MiSeq or HiSeq platform, which can produce numerous sequence reads, was applied to investigate the fecal microbiota composition in cattle. Various studies have been conducted to investigate and compare the fecal microbiota composition related to different beef production systems [10], different feed efficiencies [5,11], core microbiota investigations [12], grazing and feedlot cattle comparisons [13], management units in dairy cattle [14], dairy calves [15], and dairy cattle during early lactation [16]. These studies indicated that the fecal microbiota in cattle can be affected by various factors such as diet, age, individuals, breed, regions, and environments.
Hanwoo beef is preferred by consumers in Korea due to its higher marbling content compared to imported beef [1]. The gastrointestinal microbiota in cattle plays an important role in feed digestion, fermentation, and host health [17]. Since the composition of cattle gastrointestinal microbiota can be influenced by breed [18,19], studies on the gastrointestinal microbiota in Hanwoo cattle native to Korea will need to be conducted. Although rumen microbiota in Hanwoo cattle has been analyzed in recent studies [20,21], little research about fecal microbiota has been conducted in this breed. Different diets and genders may also affect the fecal microbiota in cattle [7,18], but there are no data to support these results in Hanwoo cattle. The goal of this study was to evaluate the impact of different diets and genders on the fecal microbiota composition in Hanwoo steers and heifers.
MATERIALS AND METHODS
A total of 44 cattle, including 22 steers and 22 heifers at the same farm, were used to evaluate the composition of fecal microbiota in Hanwoo cattle fed different diets. Fecal samples were collected by rectal grab from the 44 cattle, which were divided into four groups: (1) 11 heifers fed 45% oat hay plus a 55% TMR diet for breeding (HOTB), which included 8.6% crude protein (CP), 3.3% ether extract (EE), 52.2% neutral detergent fiber (NDF), and 27.2% acid detergent fiber (ADF), on a dry mater (DM) basis; (2) 11 heifers fed the early fattening TMR diet (HEFT), which included 13.4% CP, 5.2% EE, 51.1% NDF, and 25.9% ADF, on a DM basis; (3) 11 steers fed the early fattening TMR diet (SEFT), which included 13.4% CP, 5.2% EE, 51.1% NDF, and 25.9% ADF, on a DM basis, and; (4) 11 steers fed the late fattening TMR diet (SLFT), which included 14.5% CP, 5.2% EE, 50.5% NDF, and 25.3% ADF, on a DM basis. The TMR diet was composed of orchard grass straw and a mixture of corn germ meal, oat hull, oat hull pellets, corn cobs, palm kernels, beet pulp, barley pellets, distiller’s dried grains with solubles, corn gluten feed, lupin, molasses, and supplements. The same early fattening TMR diet was fed to steers and heifers in the SEFT and HEFT groups. Before fecal sampling, all cattle were fed these diets for more than 1 month.
Metagenomic DNA was extracted from the 44 fecal samples using the bead-beating plus column (RBB+C) method [22]. Bacterial 16S rRNA gene amplicon sequencing was conducted using the primers 341F (5′-CCTACGGGNGGCWGCAG-3’) and 805R (5′-GACTACHVGGGTATCTAATCC-3’), and the resulting amplicons were sequenced on an Illumina MiSeq platform (Illumina, San Diego, CA, USA) at Macrogen (Seoul, Korea) as described previously [20,23].
The resultant 16S amplicon sequences were cleaned and analyzed using Quantitative Insights into Microbial Ecology (QIIME 2, version 2021.8) [24] and MicrobiomeAnalyst [25] software packages. The DADA2 plugin was used to remove adapter and chimeric sequences and conduct quality filtering, denoising, and merging. Microbial diversity and taxonomy were analyzed using the cleaned amplicon sequence variants (ASVs). The ASVs were taxonomically classified against the SILVA taxonomy database (version 138). Alpha diversity indices, such as observed ASVs, Chao1, Shannon’s index, and Simpson’s index, were analyzed based on ASV biological observation matrix (BIOM) tables while the beta diversity of the fecal microbiota among the four treatment groups was analyzed using principal coordinate analysis (PCoA) based on Bray-Curtis dissimilarity matrices. Functional genetic profiles were predicted using Phylogenetic Investigation of Communities by Reconstruction of Unobserved States 2 (PICRUSt2), which was used to analyze MetaCyc pathway profiles as described previously [26]. Overall functional genetic profiles of predicted Kyoto Encyclopedia of Genes and Genomes (KEGG) orthologs were compared among the four treatment groups using principal components analysis (PCA).
Taxa and MetaCyc pathway profiles that were differentially abundant among the four treatment groups were analyzed using the linear discriminant analysis (LDA) effect size (LEfSe) (LDA score > 3) [27].
The Firmicutes/Bacteroidota (F/B) ratio and alpha diversity indices were compared among the four treatment groups using the analysis of variance (ANOVA) with Tukey’s test in the XLSTAT statistical software package (version 2022.3.1) (Addinsoft, New York, NY, USA). Beta diversity analysis and functional genetic profiles were compared among the four treatment groups using permutational multivariate analysis of variance (PERMANOVA) with PAST3 with 9999 random permutations [28].
RESULTS
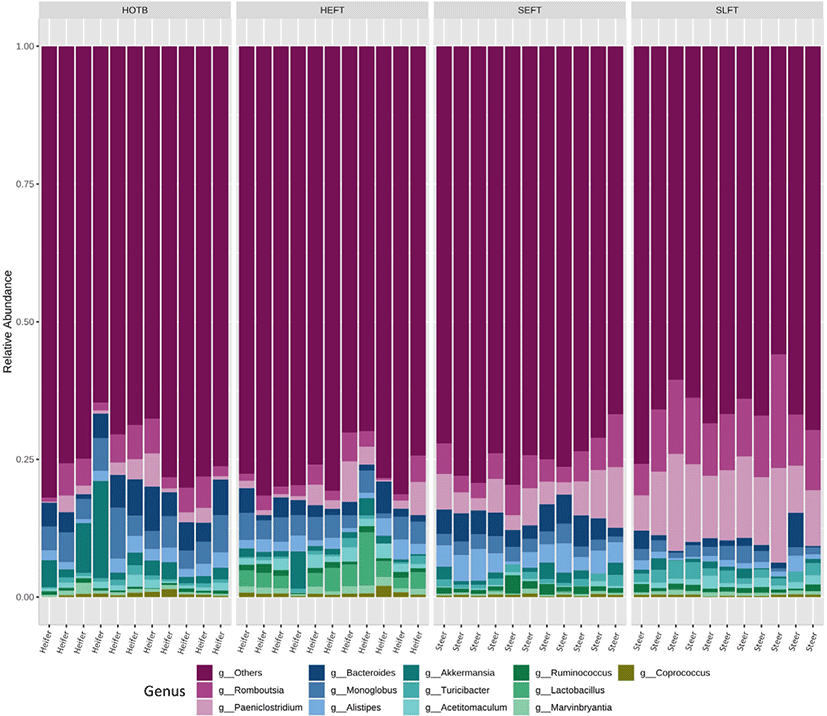
A total of 2,118,514 cleaned sequences were obtained from the 44 fecal samples, including 22 steer and 22 heifer samples. Each sample was represented by > 40,000 cleaned sequences. Good’s coverage was more than 99.8% for all fecal samples. Taxa accounting for at least 0.5% of all sequences across all 44 fecal samples were regarded as “major taxa” and used for further analysis. Firmicutes was the most dominant phylum and accounted for 75.2% of all sequences, while Bacteroidota was the second most dominant and accounted for 19.1% of all sequences (Fig. 1). Verrucomicrobiota was represented by 2.0% of the total sequences, followed by Actinobacteriota (1.1%) and Patescibacteria (1.0%) (Fig. 1). The remaining phyla accounted for < 0.5% of all sequences. At the genus level, Romboutsia was the most dominant and accounted for 5.7% of all sequences across all 44 samples, followed by Paeniclostridium (5.6%), Bacteroides (3.3%), Monoglobus (3.2%), Alistipes (2.2%), Akkermansia (2.0%), (1.1%), Acetitomaculum (1.0%), Ruminococcus (0.9%), Lactobacillus (0.9%), Marvinbryantia (0.7%), Coprococcus (0.5%), and Blautia (0.5%) (Fig. 2). The remaining genera accounted for < 0.5% of all sequences.
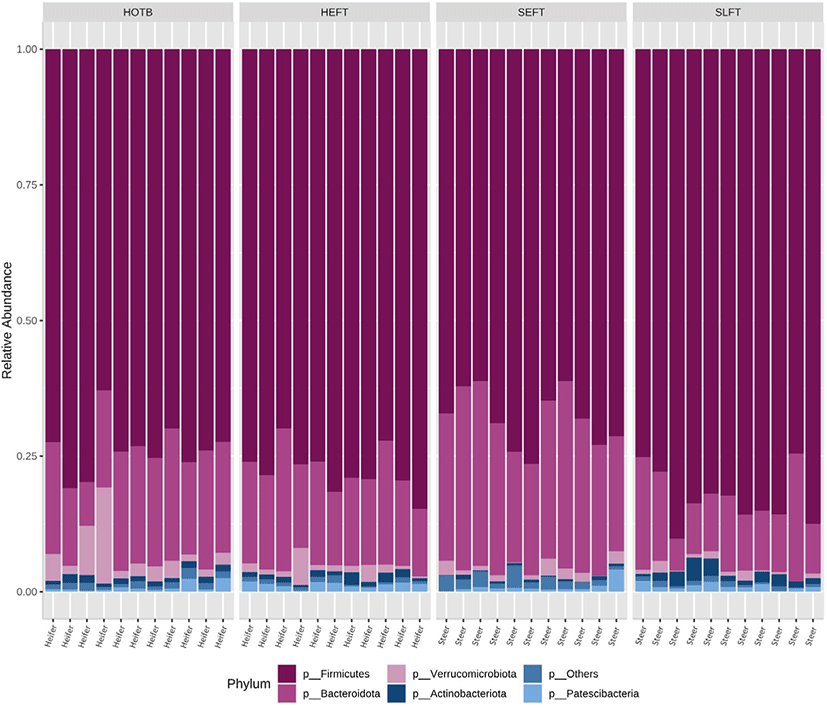
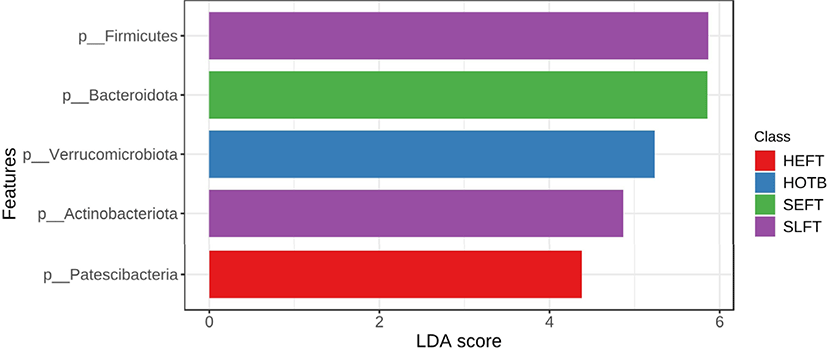
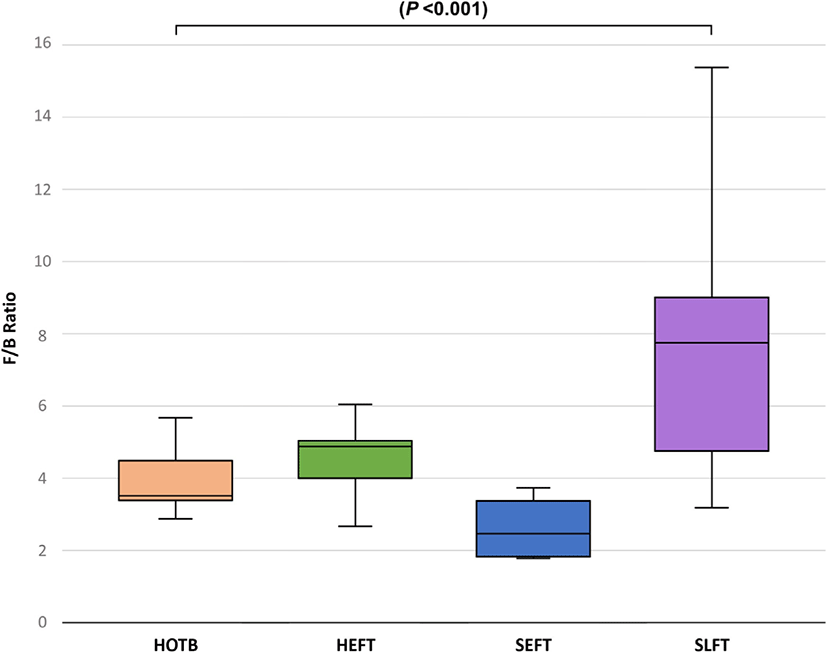
At the phylum level, LEfSe analysis indicated that Firmicutes and Actinobacteria were more abundant in the SLFT group than in the other groups, while Bacteroidota was more abundant in the SEFT group than in the other groups (Fig. 3). The F/B ratio was more abundant in the SLFT group than in the other groups (p < 0.05) (Fig. 4). Verrucomicrobiota were more abundant in the HOTB group than in the other groups, while Patescibacteria were more abundant in the HEFT group than in the other groups (Fig. 3).
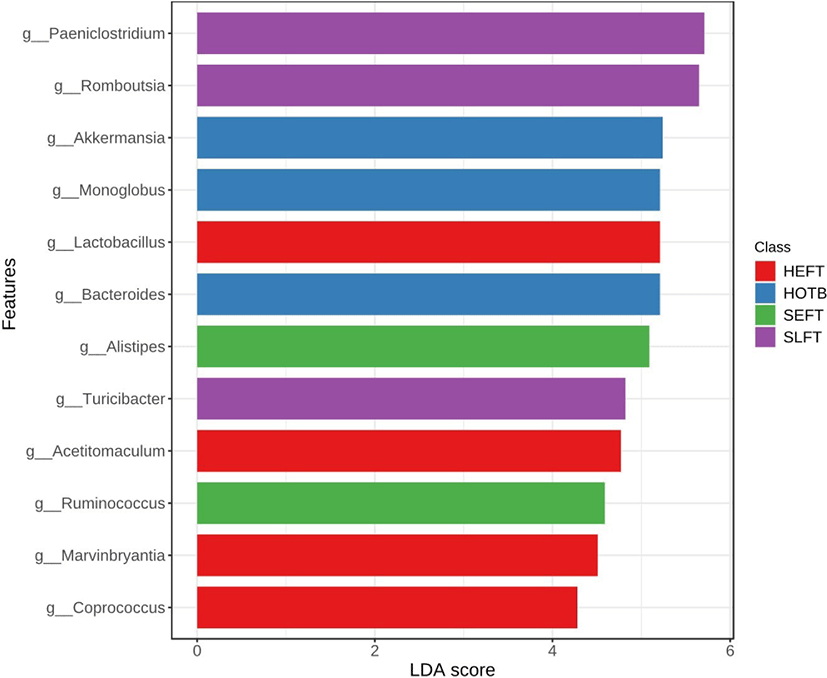
At the genus level, LEfSe analysis showed that Romboutsia, Paeniclostridium, and Turicibacter were more abundant in the SLFT group than in the other groups, while Akkermansia, Bacteroides, and Monoglobus were more abundant in the HOTB group than in the other groups (Fig. 5). Lactobacillus, Acetitomaculum, Blautia, Marvinbryantia, and Coprococcus were more abundant in the HEFT group than in the other groups (Fig. 5). In addition, Alistipes and Ruminococcus were more abundant in the SEFT group than in the other groups (Fig. 5).
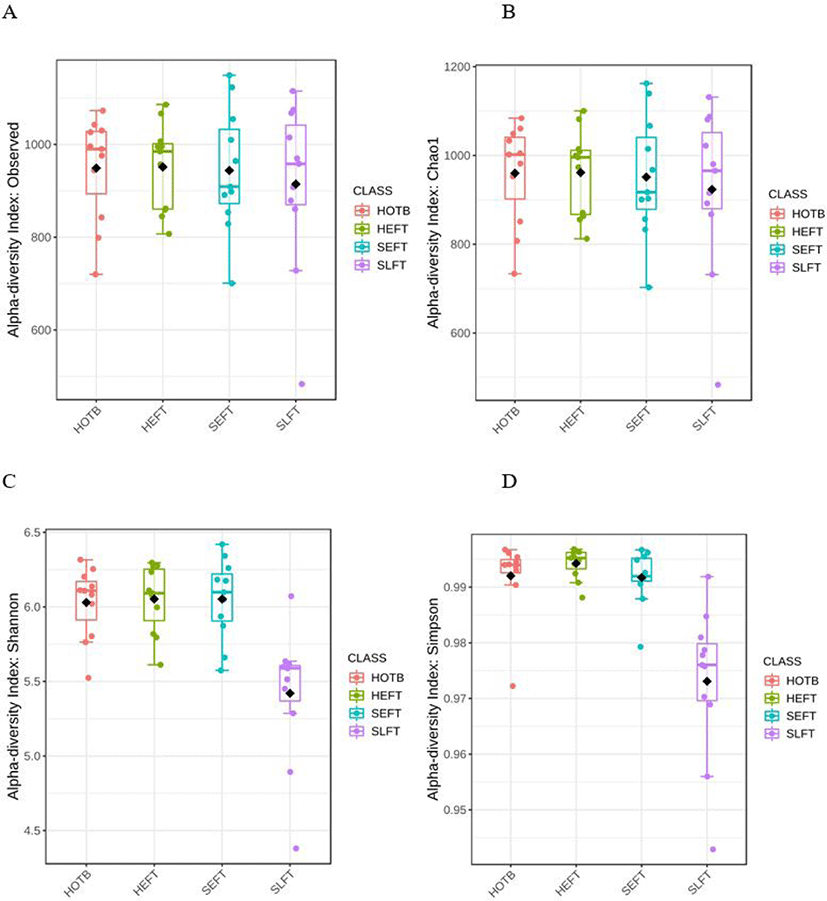
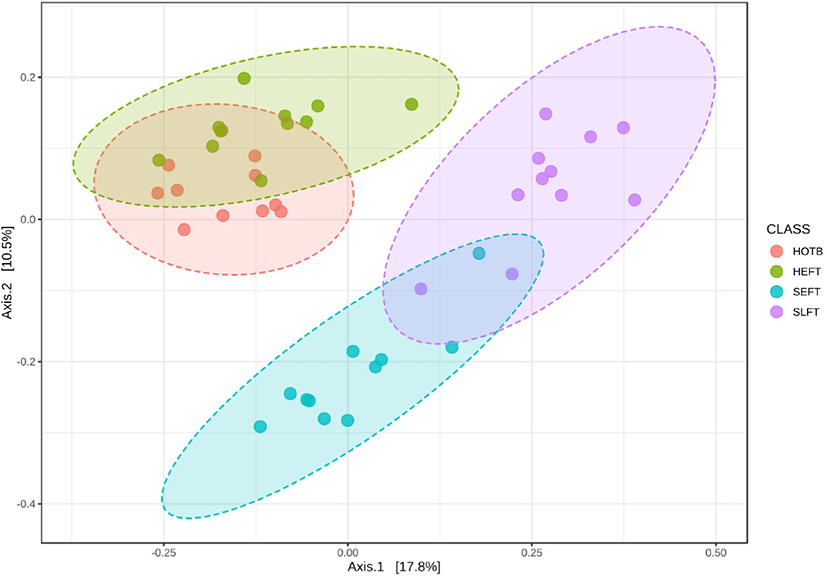
The observed ASV and Chao1 indices did not differ (p > 0.05) among the four treatment groups (Figs. 6A and 6B), whereas Shannon and Simpson diversity indices were lower (p < 0.05) in the SLFT group than in the other groups (Figs. 6C and 6B). Beta diversity analysis based on Bray-Curtis dissimilarity matrices showed that overall fecal microbiota differed (p < 0.05) not only among the four treatment groups, but also between different gender groups (Fig. 7).
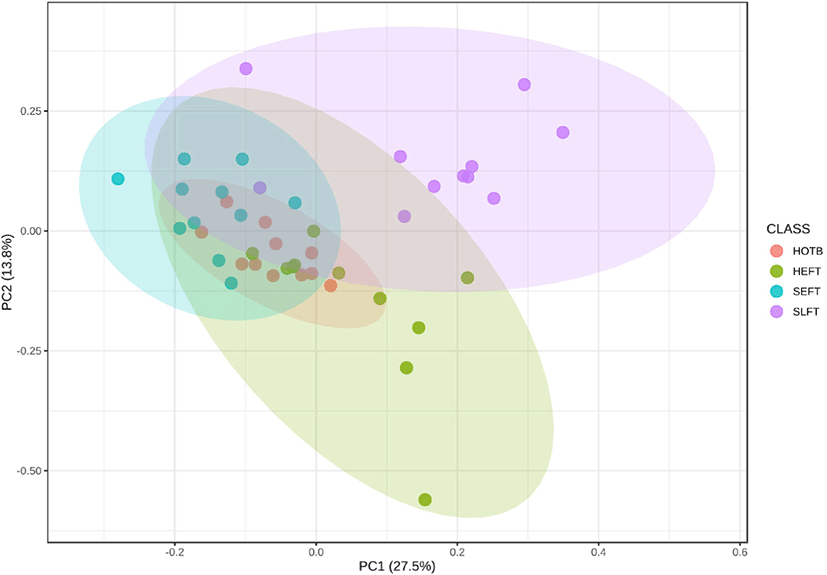
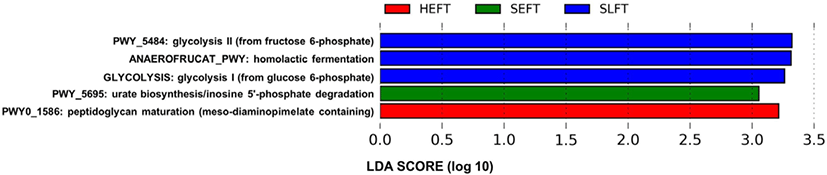
The PCA plot revealed that the distribution of functional genetic profiles differed (p < 0.05) among the four treatment groups based on PERMANOVA (Fig. 8). LEfSe analysis indicated that the MetaCyc pathways related to glycolysis (PWY_5484 and GLYCOLYSIS) and homolactic fermentation (ANAEROFRUCAT_PWY) were more predominant in the SLFT group than in the other groups, while the MetaCyc pathways related to peptidoglycan maturation (PWY0_1586) were more predominant in the HTEF group than in the other groups (Fig. 9). In addition, the MetaCyc pathway related to urate biosynthesis/inosine 5’-phosphate degradation (PWY_5695) was more predominant in the SEFT group than in the other groups (Fig. 9).
DISCUSSION
Fecal microbiota influences the productivity, health, and well-being of cattle [4,7]. Previous studies revealed that rumen microbiota can be affected by various factors, such as diets, hosts, breeds, geographic regions, and seasons [18,19]. The fecal microbiota of cattle can also be affected by these factors, particularly diet [7]. However, to date, there has been no study to evaluate the impact of different diets on fecal microbiota in Korean native Hanwoo steers and heifers. Thus, we investigated the impact of different diets on the composition of fecal microbiota in Hanwoo steers and heifers at the same farm. We also examined the impact of different genders on the fecal microbiota composition in Hanwoo steers and heifers fed the same diet at the same farm.
Different diets seem to affect the composition of the fecal microbiota in both Hanwoo steers and heifers based on beta diversity analysis. Previous studies showed that both rumen and fecal microbiota were greatly affected by different diets [7,18]. In the Hanwoo heifers in this study, the high forage contained in the diet for breeding may result in changes in the fecal microbiota composition compared to the that of heifers fed the early fattening diet. In Hanwoo steers, the slightly higher concentrate contained in the late fattening diet may contribute to shifts in the fecal microbiota composition compared to that of steers fed the early fattening diet. Otherwise, since the feed ingredients and chemical composition were not very different between the early and late fattening diets, the different cattle ages may have influenced differences in the fecal microbiota composition. In addition, different genders seem to influence the composition of the fecal microbiota in Hanwoo cattle, as shown in the rumen [18]. Although the same early fattening TMR diet was fed to steers and heifers at the same farm, the overall microbial composition was distinct between steers and heifers based on beta diversity analysis. This microbial difference seems to result from different genders in Hanwoo cattle.
The F/B ratio in the rumen is positively correlated with the average daily gain in ruminants [29]. The present study showed that the F/B ratio in the feces was the greatest in late fattening steers. Since steers have a greater average daily gain compared to heifers [30], a greater F/B ratio in the feces of Hanwoo steers at the late fattening stage may be associated with a high feed efficiency, as shown in the rumen. This may be due to differences in host genetics between steers and heifers. Thus, host genetics and fecal microbiota may jointly contribute to feed efficiency in cattle [31]. Further studies are required to examine host genetics and fecal microbiota affecting feed efficiency.
Paeniclostridium and Romboutsia utilize various organic substrates [32,33] and both genera were dominant and more abundant in the feces of Angus cattle fed the TMR diet than in grazing cattle [13]. Similarly, both Paeniclostridium and Romboutsia were dominant in Hanwoo steers fed the TMR diet, particularly in the late fattening Hanwoo steers. In late fattening steers fed a high grain diet, undigested carbohydrates in the large intestine may be increased and may lead to an increase in Paeniclostridium and Romboutsia utilizing various organic substrates. Otherwise, the older age of the late fattening steers may affect the fecal microbiota composition. Interestingly, heifers had a lower Paeniclostridium abundance in the feces, although the same early fattening TMR diet was fed to both steers and heifers, thus indicating that different genders may affect the abundance of Paeniclostridium. Akkermansia was typically found in bovine feces and was abundant in cattle fed a high forage diet, resulting in the degradation of the mucus layer, as described in previous studies [7,13,34]. Previous studies indicated that the abundance of Bacteroides was low in cattle fed a low forage diet [7]. However, Bacteroides was abundant in Hanwoo heifers fed the high forage diet for breeding, indicating that the fecal microbiota may be influenced by different breeds or environments. Monoglobus can ferment pectins [35] and the high abundance of Monoglobus in Hanwoo heifers fed a high forage diet for breeding may be due to pectins contained in the undigested forage in the large intestine. Alistipes is a core genus that is commonly found in the feces of cattle, but its function requires further investigation [7,16,36]. Previous studies showed that Turicibacter is more abundant in cattle with a high feed efficiency [11,37]. Since steers have a greater average daily gain compared to heifers, the abundance of Turicibacter is thought to be the greatest in Hanwoo steers fed a late fattening diet in the present study. Acetate-producing Acetitomaculum, which can utilize formate and glucose, was isolated from bovine rumen [38], and may also be present in the feces of cattle. However, further studies must be conducted to verify this assumption. Lactobacillus and Blautia have probiotic characteristics [39] and were more abundant in Hanwoo heifers than in Hanwoo steers, and thus may be affected by different genders. Blautia is less abundant in cattle fed a high forage diet compared to cattle fed a high grain diet [7]. Similarly, the abundance of Blautia was lower in heifers fed a high forage diet for breeding than in those fed the late fattening diet including high concentrate. Marvinbryantia, which is present in the human gut and has the ability to degrade cellulose [40], was also found in the feces of cattle [41]. Since Marvinbryantia is more abundant in early fattening heifers than in early fattening steers, this genus may be affected by gender. A previous study showed that the abundance of Coprococcus was low in heifers fed a high forage diet [7]. However, in the present study, the abundance of Coprococcus was high in Hanwoo heifers fed a high forage diet. Because Coprococcus does not seem to be affected by the ratio of forage and concentrate, it may be influenced by different breeds or environments.
Hao et al. [42] compared differences in rumen microbiota composition based on age and found that older cattle had low alpha diversity indices. In the present study, low Shannon and Simpson diversity indices were the lowest in the feces of Hanwoo steers at the late fattening stage, indicating that alpha diversity may be affected by age, as shown in the rumen. Further studies will need to be conducted to identify the association between age and fecal microbiota in cattle.
The increase in the functional genetic profiles related to glycolysis in steers fed the late fattening TMR diet may be associated with the F/B ratio increase. In addition, the increase in the F/B ratio may be attributed to increased glycolysis and fermentation by various bacteria in the Firmicutes phylum. Since Paeniclostridium, Romboutsia, and Turicibacter belong to the Firmicutes phylum, a higher abundance of these genera in steers fed the late fattening TMR diet may contribute to the F/B ratio increase, which is positively correlated with the average daily gain [29]. This is supported by the association between Turicibacter and a high feed efficiency [11,37]. The increase in many minor bacterial species within Firmicutes may also contribute to the F/B ratio increase. In addition, the increases in the functional genetic profiles related to homolactic fermentation in steers fed the late fattening TMR diet may be associated with an increase in lactic acid-producing bacteria undergoing homolactic fermentation. Since homolactic bacteria, such as Pediococcus, Streptococcus, Lactococcus, Enterococcus, and Lactobacillus, belong to the Firmicutes phylum [43], the increase in homolactic bacteria may also contribute to the F/B ratio increase in steers fed the late fattening TMR diet. Further studies will need to be conducted to confirm associations between the F/B ratio and bovine fecal bacteria, including lactic acid-producing bacteria.
The results of this study suggest that fecal microbiota is affected by different diets and genders in Hanwoo cattle. The F/B ratio associated with high feed efficiency was greater in steers than in heifers, while bacterial diversity seemed to be affected by a high concentrate diet. The increase in MetaCyc pathways related to glycolysis and homolactic fermentation may be associated with the F/B ratio increase. In future studies, the use of an appropriate diet considering gender may be important to maintain bovine gut health. In addition, further studies should be performed to determine if increasing the F/B ratio in the fecal microbiome can be used as a potential strategy to improve the feed efficiency of Hanwoo cattle.