INTRODUCTION
Duck is the second most frequently consumed type of poultry in Asia. The improvement of carcass characteristics and meat quality is beneficial to consumers. High intramuscular fat (IMF) in duck pectoral muscle is gradually gaining popularity among consumers and researchers in the poultry industry [1]. IMF mainly accumulates in muscle cells [2] and affects meat duck flavour, tenderness, and quality [3,4]. Lipids include triglycerides (TGs), phospholipids, and cholesterol (CHOL), which are insoluble in water, and their transport in the extracellular fluid, lymph, and blood requires proteins associated with the lipids to suspend them in water [5]. Therefore, IMF is a key indicator for evaluating duck meat quality and it is affected by proteins that interact with lipids.
Apolipoprotein H (APOH) binds to negatively charged phospholipids located in the cell core and attaches to the partial structure of various lipoproteins [6]. APOH is involved in several human physiological processes, such as lipid metabolism, proliferation, inflammation, thrombosis, systemic lupus erythematosus, cognitive aging, metabolism of TG in protein lipoprotein lipase (LPL), and participation in the immune response process of anti-phospholipid syndrome (APS) [7–14]. LPL has important functions in lipid metabolism, such as participation in fatty acid transport and TG hydrolysis [15,16]. APOH has been reported to inhibit peroxisome proliferator activated receptor gamma (PPARG) expression and increase LPL activity in macrophages[17–19]. PPARG has positive effects not only in regulating cellular lipid metabolism but also in cytokine production and inflammation inhibition [20]. Apolipoprotein plays an essential role in the two stages of chicken skeletal muscle myogenesis and adipogenesis [2], and the PPAR pathway is involved in lipid deposition in chicken skeletal muscle [21]. However, few studies have addressed the mechanisms involved in regulating lipid metabolism in duck myoblasts.
A previous study showed that APOH is the difference gene between high and low groups of meat ducks with residual feed intake, and the factor affecting the residual feed intake of meat ducks is lipid metabolism [22]. Therefore, the APOH gene may play an important role in duck lipid metabolism. To clarify the key role of APOH gene in the lipid metabolism of duck myoblasts, this study constructed a cell model of APOH gene knockdown and overexpression and detected the intracellular conditional factors and indicators for analysis. This study provides a molecular basis for breeding ducks with high IMF by explaining the molecular mechanism of APOH gene promoting fat accumulation in duck myoblasts.
MATERIALS AND METHODS
The animal experiment was reviewed and approved by the Institutional Animal Care and Use Committee of Anhui Agriculture University (no. SYDW-P20190600601). Duck myoblasts were isolated from thoracic tissue of 13 embryonic-day-old duck embryos. Duck embryos are provided by Qiangying Duck Industry, Anhui.
The breast tissues of 12 duck embryos that had been hatched for 13 days were removed, cut into pieces, added with five times the volume of type I collagenase and digested in a 37°C water bath for 2 h. After digestion until there is no large tissue clumps, a equal volume of Dulbecco’s Modified Eagle Medium (DMEM)/F12 (Biological Industries, Kibbutz Beit-Haemek, Israel) was added to terminate digestion. Collect the digested tissue supernatant, filter it through a 300-mesh filter, and collect the filtrate. The filtrate was collected into a 15 mL centrifuge tube, centrifuged at 1,000 rpm/min for 10 min, collect the cell precipitation, 2 mL of culture medium was added to resuspend the cells, and the cells were collected by centrifugation again under the same conditions. Ten millilitres of complete medium (79% DMEM/F12 medium + 20% fetal bovine serum [FBS; Biological Industries] + 1% antibiotics [Thermo Fisher Scientific, Waltham, MA, USA]) was added to the cells (CS) to resuspend and seed them in a 10 cm petri dish and then they were incubated at 37°C for 4 h.
A large number of myoblasts (CS1) and a small number of other cells were in a nonadherent state. To obtain purer myoblasts, the culture of the supernatant from the first culture dish was repeated and cultured for 4 h. Finally, the supernatant from the second dish was collected, the number of cells (CS2s) was recorded and they were seeded in 6-well plates. Medium containing 89% DMEM/F12, 10% FBS and 1% antibiotics was used to culture the CS. The incubator conditions were 37°C and 5% CO2.
When cells grew to 80% confluence, they were reseeded into 24-well plates for immunofluorescence staining of PAX7 (Proteintech, Wuhan, China) [22,23]. The cells were fixed for 15 min with 4% paraformaldehyde (Biosharp, Hefei, China), blocked, and incubated with anti-paired box (PAX7) primary antibody at 4°C overnight. The anti- PAX7 was detected with a secondary antibody labeled with fluorescein isothiocyanate (FITC) (Immunoway, Plano, TX, USA) in a darkroom at 25°C for 1 h. Finally, the cells were incubated with 4′,6-diamidino-2-phenylindole (DAPI) (Servicebio, Wuhan, China) in the darkroom for 15 min. Then the immunofluorescence images of PAX7 protein labeled were observed by fluorescence microscope (DP73, Olympus, Tokyo, Japan).
The duck APOH (XM_005016097.5) coding region was amplified with forward primer 5`- GGATCCATGTACTCCCTGGTGCTGGTCG -3` (contain BamHI restriction site) and reverse primer 5`-GCTAGCTCATTCATGATCTTCACATGGTTTC-3` (contain NheI restriction site). Polymerase chain reaction (PCR) (T100, Bio-Rad, Hercules, CA, USA) was used to amplify a 1065 bp product and it was purified. The APOH sequence and pBI-CMV3 vector containing BamHI (NEB, Beijing, China) and NheI (NEB) restriction endonuclease cleavage sites were digested and ligated together (a gift from Zhao Zhihui’s research group at Guangdong Ocean University). The plasmid pBI-CMV3-APOH was double-enzyme digested and confirmed by agarose gel electrophoresis. Shanghai Sangon Biotechnology Co., Ltd. sequenced the plasmid to validate it.
APOH interfering RNA (shRNA) was designed according to its mRNA sequence (XM_005016097.5) with online software (https://www.thermofiser.com/us/en/home/brands/invitrogen/ambion.html), and APOH-shRNA interference sequences (5`-AGAGGTCTTCG CCACAATCAATGTTCAAGAGACATTGATTGTGGCGAAGAGCTTTTTTG-3` and 5`- GATCCAAAAAAGCTCTTCGCCACAATCAATGTCTCTTGAACATT GATTGTGGCGAAGAGC-3`) were synthesized. The shRNA annealed product was purified and ligated into pGPU6-GFP-Neo knockdown vector (a gift from Zhao Zhihui Research Group at Guangdong Ocean University).
When the cells grew to 70%–80% confluence, we transfected them with the overexpression, knockdown, and normal control plasmids of APOH with Exfect, with at least three replicates per treatment. After 24 h of transfection, the corresponding experiment was carried out.
Then, total RNA was extracted by TRIzol (Thermo Fisher Scientific). cDNA (1 μg) was reverse transcribed using an RT-cDNA synthesis kit (Vazyme, Nanjing, China). PCR mix (Vazyme) was used in the real-time PCR assay. The reaction system was 95°C for 5 min, 95°C for 30 s, and 60°C for 30 s for 35 cycles. With glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as the reference gene, quantitative PCR was used to detect the gene’s mRNA level. Shanghai Shenggong Biotechnology Co., Ltd. synthesized the RT-PCR primers (Table 1). GAPDH was used as the reference gene, and an experiment was performed in triplicate. The relative expression of genes was calculated using 2−ΔΔCt.
1) qRT-PCR, quantitative real-time polymerase chain reaction; bp, base pair; APOH, apolipoprotein H; AKT1, AKT serine/threonine kinase 1; AMPK, AMP-activated catalytic subunit alpha 1; ACC1, acetyl-CoA carboxylase 1; PPARG, peroxisome proliferator activated receptor gamma; ACSL1, acyl-CoA synthetase long chain family member 1; ELOVL6, ELOVL fatty acid elongase 6; FAS, fas cell surface death receptor; LPL, lipoprotein lipase; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Cells were harvested 24 hours after transfection of the plasmid. After aspirating the medium, the cells were scraped off with a cell scraper and collected into a centrifuge tube after washing with phosphate-buffered saline (PBS). The centrifuge tube containing cells was centrifuged, the supernatant was removed, RIPA protein lysis buffer and protease inhibitors were added, and the cells were lysed for 10 min on ice. The lysed cells were centrifuged at 13,000 rpm/min (4°C) for 10 minutes to obtain a supernatant containing total protein. To keep the sample loading consistent, bicinchoninic acid (BCA) kits were used for protein concentration detection and quantification of total protein. For western blotting (PowerPac™ HV, Bio-Rad), consistent amounts of protein were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then the total protein was transferred to polyvinylidene difluoride (PVDF) (88518, Thermo Fisher Scientific) membranes. PVDF was incubated with 1% bovine serum albumin (BSA) for 2 h at room temperature and washed with tris-buffered saline (TBST). Then, the membrane was incubated with primary anti-APOH (1:1,000) antibody overnight at 4°C. Primary antibodies (ACC1, ACSL1, PPARG, LPL, ELOVL6, AKT, Pakt, AMPK, pAMPK) were diluted at a dilution ratio of 1:1,000. After the primary antibody was incubated and washed three times with TBST, the anti-rabbit secondary antibody (Immunoway) was incubated for 2 h. Enhanced chemiluminescence horseradish peroxidase (HRP) substrate was used to observe the stained protein bands. After taking an image with a chemiluminescence instrument, ImageJ was used to scan the gray value of the proteins.
Oil red O stock solution and working solution was prepared according to the previous research method [23].
After 24 h of transfection, the cells were washed with PBS after aspirating the cell culture medium. Cells were incubated with 4% paraformaldehyde (Biosharp, Hefei, China) for 30 min and washed three times with PBS. The washed cells were stained with Oil red O working solution at room temperature for 30 min, the Oil red O working solution was removed, 60% isopropanol was used to wash the cells three times, and finally the cells were washed three times with PBS. Then, 200 µL of 60% isopropanol was added to the cells for 20 min, and a microplate reader was used to measure the absorbance at 510 nm.
According to the kit instructions, the TG (Applygen, Beijing, China) and CHOL (Applygen, China) contents in CS2s were detected. The CS2 cells were collected 24 h after transfection with the plasmids. Cell numbers of different samples were unified by BCA protein quantification and three experiments were repeated for each model. TG and CHOL in the cell were normalized to the protein content. Use a microplate reader to measure the absorbance of different samples at a specific absorbance, and calculate the content of TG and CHOL.
RESULTS
After 24 h, the cells began to adhere to the wall and became spindle cells. Most of the CS2s extended at both ends after culturing for 36 h. After 48 h, they entered the logarithmic growth phase (Figs. 1A–1C). The cells were identified with the CS2-specific marker PAX7 for immunofluorescence identification (Figs. 1D–1F).

The vector of overexpression (pBI-CMV3-APOH) and knockdown (pGPU6-GFP-Neo-APOH) APOH gene were constructed to examine the function of APOH in CS2s. The restriction enzyme digestion verification (Fig. 2C) shows that we successfully cloned the 1,053 bp duck APOH and constructed a recombinant expression vector. Sequencing technology confirmed that the interference target sequence was successfully recombined into pGPU6-GFP-Neo (Fig. 2D).
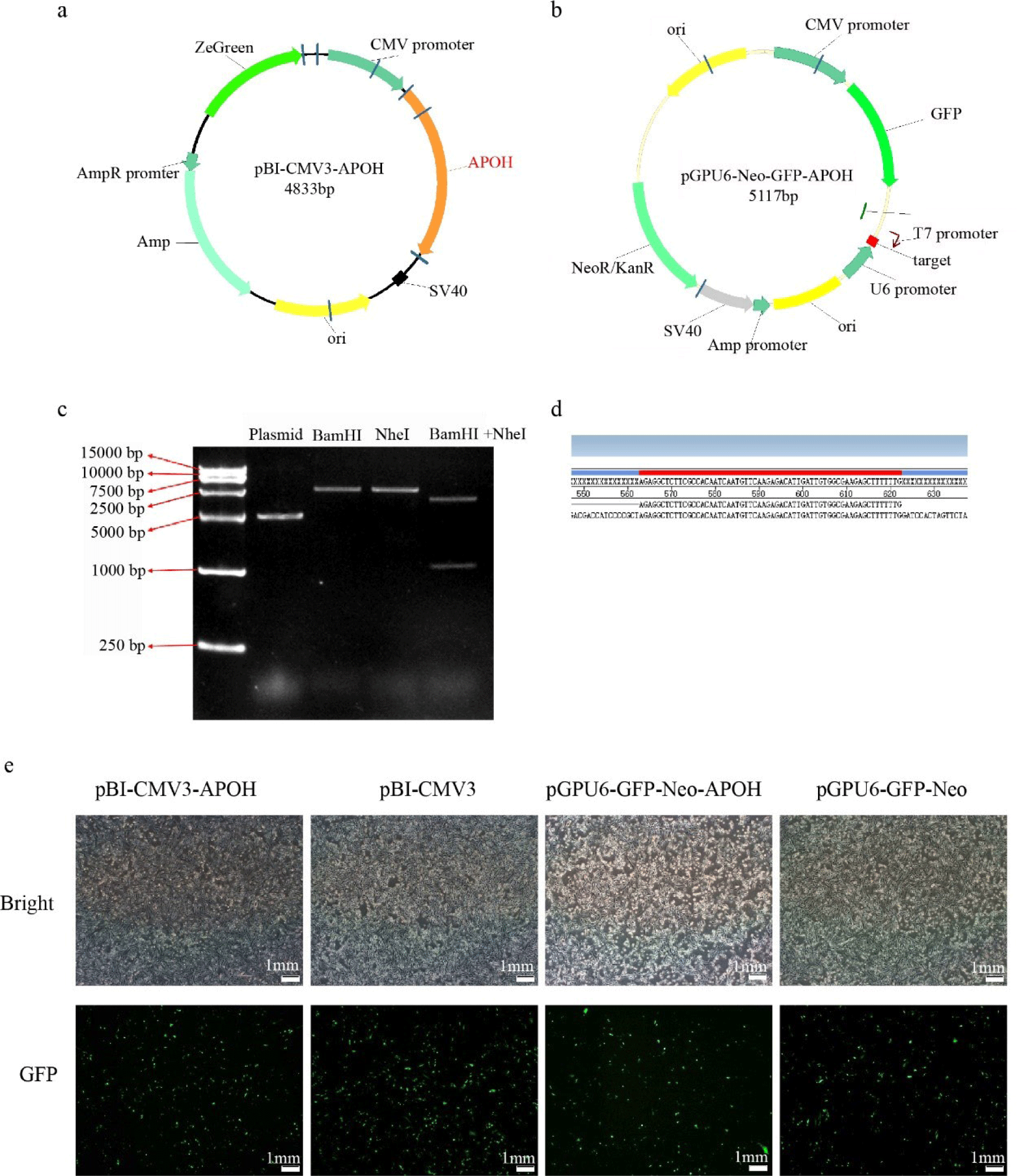
After successful construction, the pBI-CMV3-APOH, pBI-CMV3, pGPU6-GFP-Neo-APOH, and pGPU6-GFP-Neo vectors were transfected into the CS2s when they attained 80% confluency. After 24 h of transfection, green fluorescent proteins were observed in the cells under an inverted fluorescence microscope (Fig. 2E). The results showed that the APOH gene overexpression and knockout myoblast model was successfully constructed.
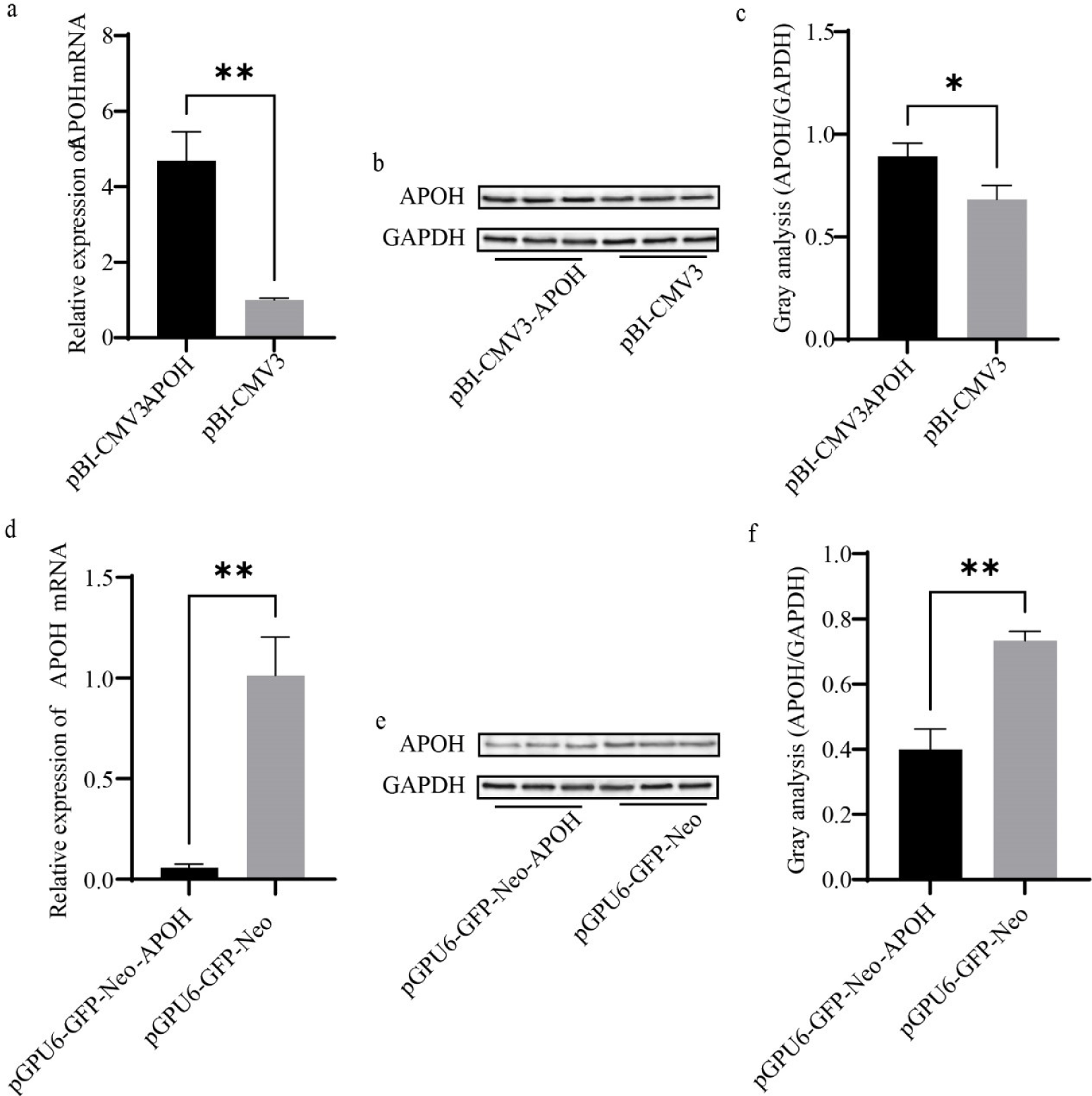
After transfection with pBI-CMV3-APOH, pGPU6-GFP-Neo-APOH, and the corresponding empty vector for 24 h, the cells were collected, and the total RNA was extracted. RT-PCR and western blot were used to detect the mRNA and protein expressions of APOH in CS2s. The results showed that the mRNA expression of APOH gene in cells transfected with pBI-CMV3-APOH was significantly increased; the mRNA expression of APOH gene transfected with pGPU6-GFP-Neo-APOH was significantly lower than that transfected with empty vector (Figs. 3A and 3D). In CS2s transfected with pBI-CMV3-APOH, APOH protein overexpression was confirmed by western blot analysis (Figs. 3B and 3C). However, after pGPU6-GFP-Neo-APOH transfection, APOH protein expression was significantly reduced in CS2. (Figs. 3E and 3F).
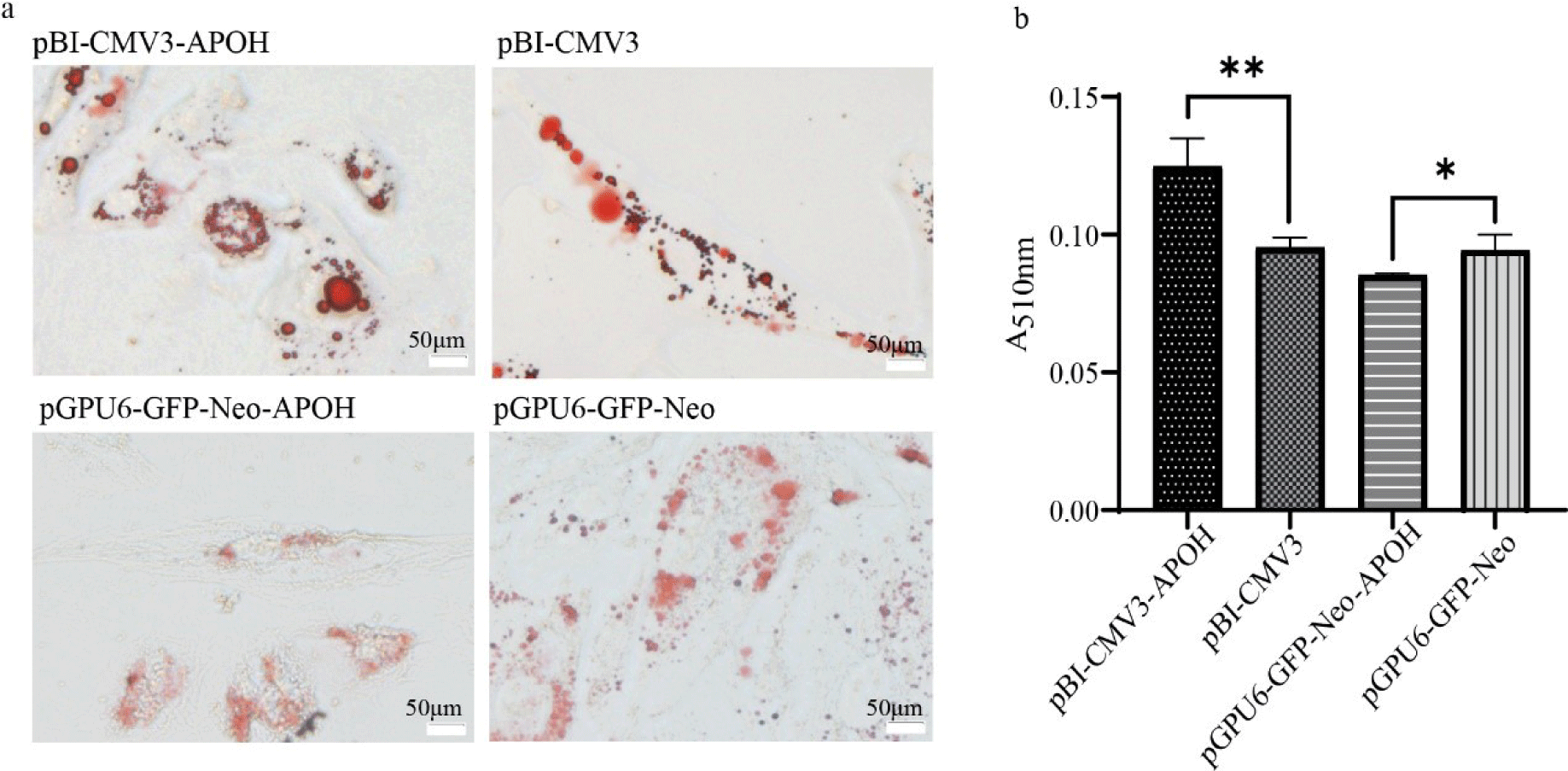
Oil red O staining was observed under an optical microscope showed that a large number of red lipid droplets appeared in CS2s of the pBI-CMV3-APOH group, and the lipid droplet area was larger than that of the pBI-CMV3 group, while a small number of red lipid droplets appeared in the CS2s of the pGPU6-GFP-Neo-APOH group, and most of them were in a diffuse state (Fig. 4A). Further lipid content determination results showed that the lipid content of the pBI-CMV3-APOH group was significantly higher than that of pBI-CMV3 group, while the lipid content was significantly reduced after knockdown of the of the APOH gene in cells (Fig. 4B).
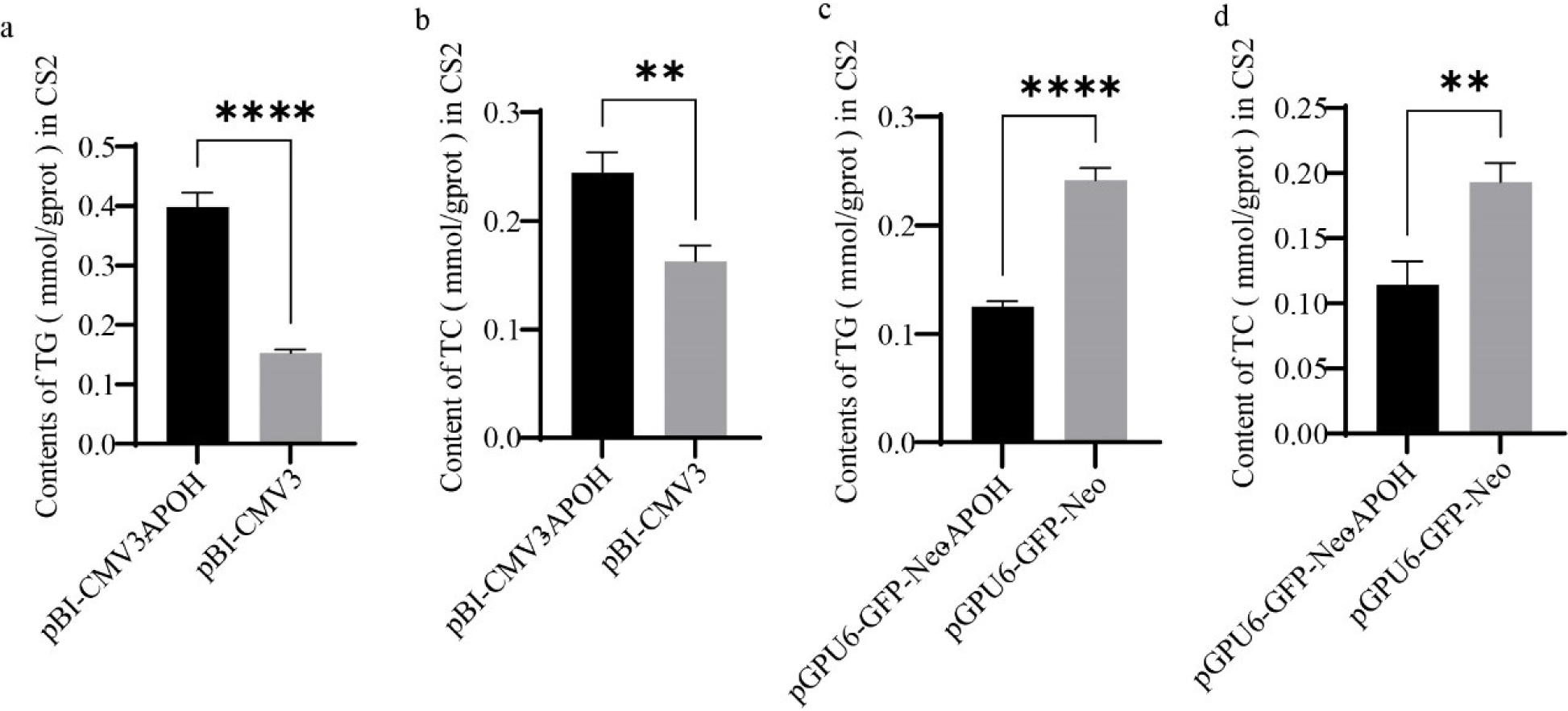
We examined the effect of APOH on the synthesis of TG in CS2s by detecting the content of TG in CS2s. Levels of TG were higher in CS2s transfected with pBI-CMV3-APOH than in those transfected with the pBI-CMV3 vector (Fig. 5A). The TG content was lower in CS2s transfected with pGPU6-GFP-Neo-APOH than in the control group, which was transfected with only the pGPU6-GFP-Neo vector (Fig. 5C). Levels of CHOL were detected with a CHOL kit. The results showed that CHOL content was higher in CS2s transfected with pBI-CMV3-APOH than in the pBI-CMV3 group (Fig. 5B) and CHOL content was lower in CS2s transfected with pGPU6-GFP-Neo-APOH than in the pGPU6-GFP-Neo group (Fig. 5D).
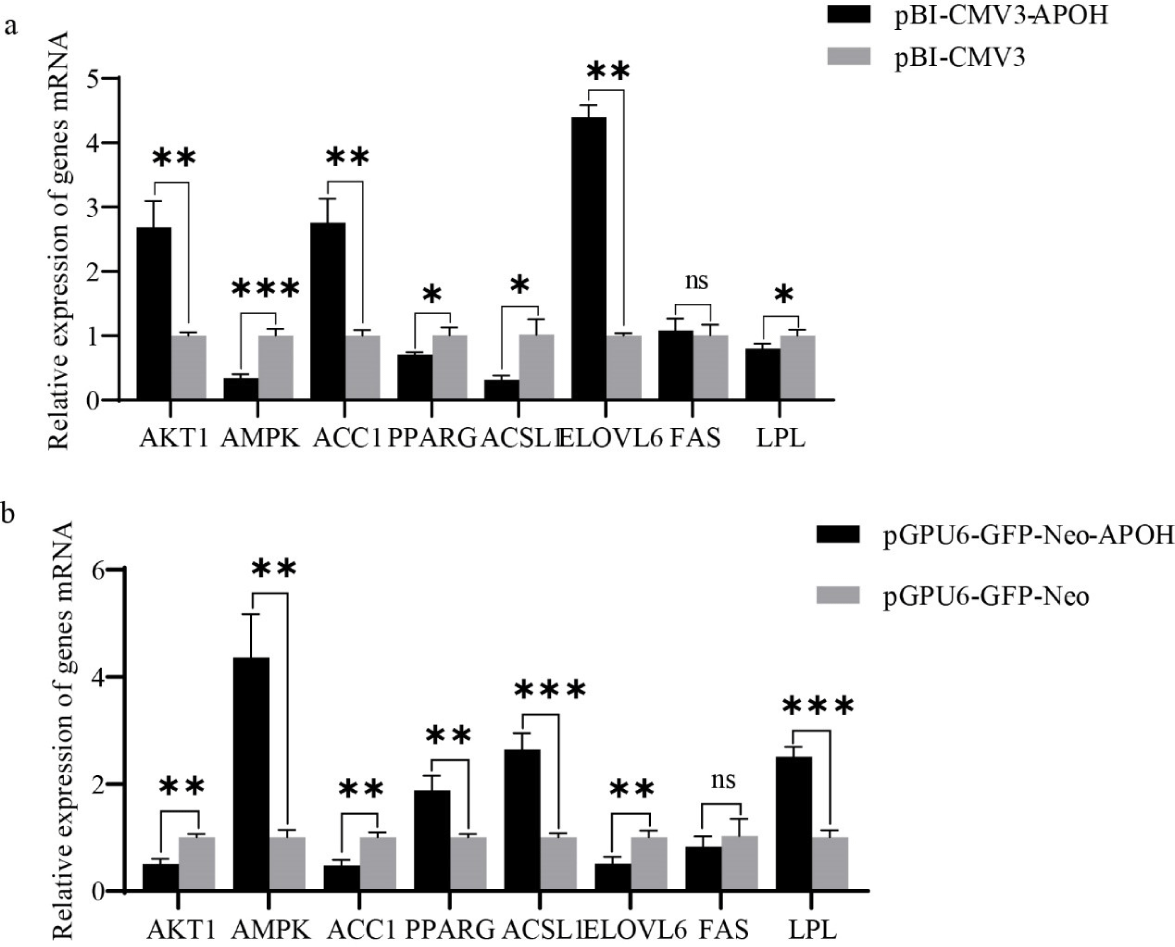
After transfection with plasmids for 24 h, the mRNA and protein of lipid metabolism-related genes in CS2 cells were detected. The results showed that compared with the control group, AKT1, ACC1 and ELOVL6 genes were significantly up-regulated after overexpression of APOH gene in myoblasts, fas cell surface death receptor (FAS) gene had no significant change, while AMPK, PPARG, ACSL1 and LPL genes were significantly down-regulated. (Fig. 6A). A contrary result was obtained when CS2s were transfected with pGPU6-GFP-Neo-APOH. When CS2s were transfected with pGPU6-GFP-Neo-APOH, the genes involved in lipid metabolism including AKT1, ACC1 and ELOVL6 were significantly down-regulated, the expression of FAS had no significant change, and the gene expression of AMPK, PPARG, ACSL1 and LPL was significantly increased (Fig. 6B). The above results indicate that APOH gene plays an important role in the lipid metabolism mechanism of ducks.
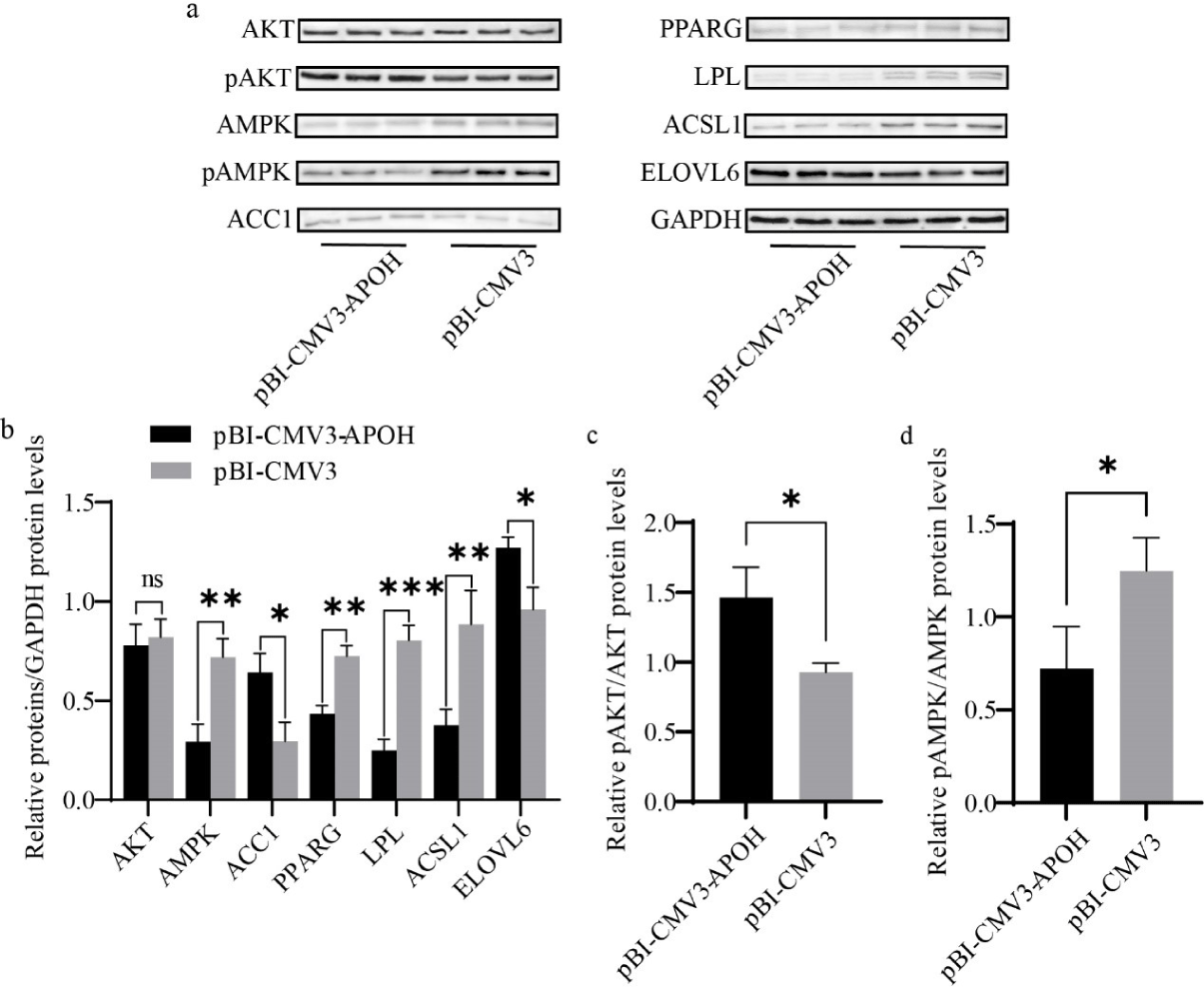
Western blotting was used to evaluate the effect of APOH gene on the protein expression levels of lipid metabolism genes in CS2s. Overexpression of APOH increased the protein expression of ACC1 and ELOVL6 and decreased AMPK, PPARG, ACSL1, and LPL, but there was no significant difference in the total protein content of AKT (Figs. 7A and 7B). Overexpression of APOH increased the phosphorylation of AKT protein and simultaneously decreased the phosphorylation of AMPK protein (Figs. 7A, 7C, and 7D).
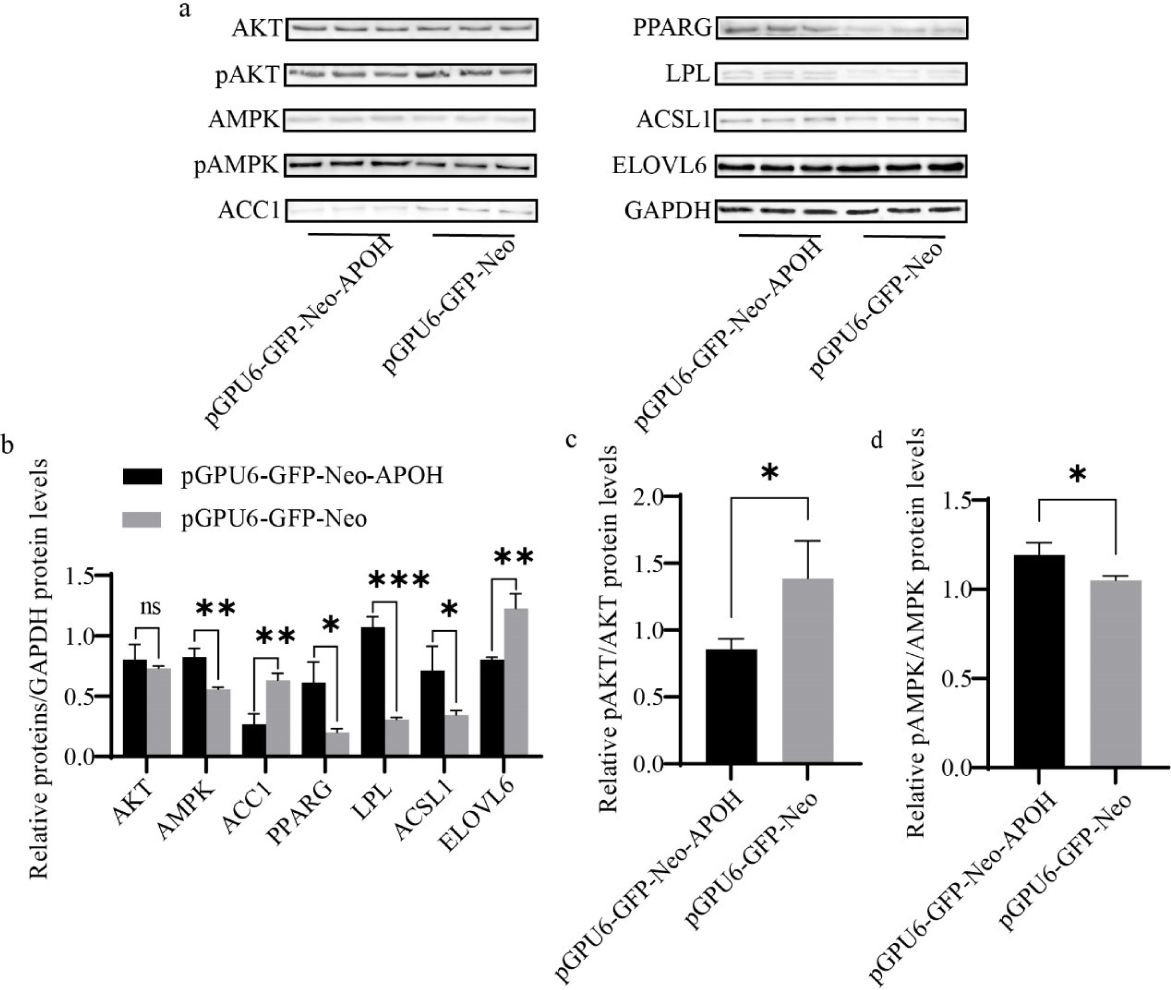
Knockdown of APOH decreased the protein expression of ACC1 and ELOVL6 and increased AMPK, PPARG, ACSL1, and LPL, but there was no significant difference in the total protein content of AKT (Figs. 8A and 8B). Knockdown of APOH decreased the phosphorylation of AKT protein and simultaneously increased the phosphorylation of AMPK protein (Figs. 8A, 8C, and 8D).
DISCUSSION
Meat duck skeletal muscle tissue is an important place for the utilization and oxidation of glucose and fatty acids and an essential tissue for maintaining blood sugar balance. Although the lipid content in skeletal muscle cells is low, skeletal muscle cells can accumulate fat and are central to fat metabolism [24]. The changes in fat metabolism affect physiological processes such as cell growth and differentiation [25]. Among these, the synthesis of TGs is a vital process of the cell. Insulin resistance and type 2 diabetes in skeletal muscle are closely related to the accumulation of TGs in skeletal muscle [26,27]. A short-term high-fat overfeeding (HFO) diet has the most significant increase in methylation of human myoblasts, including the APOH gene, and DNA methylation can affect gene expression and chromosome stability, which may affect the phenotypic results of health and disease[28]. The lower abundance of APOH gene in chicken breast muscle indicates decreased of IMF content, and the mechanism is not clear[29, 30]. APOH is one of the key proteins in lipid metabolism. How does APOH regulate the synthesis and metabolism of lipids in myoblasts to avoid lipid accumulation and prevent lipotoxicity? The study of lipid metabolism in myoblasts is significant, so we used duck myoblasts as a lipid metabolism model for meat ducks.
Previous studies have shown a positive correlation between APOH and TG and CHOL [31]. This adds favourable evidence to our research results and indicates that APOH affects the TG and CHOL contents in myoblasts. Overexpression of APOH in myoblasts (CS2s) increased the contents of TG and CHOL. Furthermore, the inhibition of APOH expression in CS2s suppressed the TG and CHOL contents. The homeostasis of lipid metabolism throughout the nucleus depends on the regulation of these signals to form both complex and elaborate signal transduction pathways.
ACC1 is a multifunctional enzyme containing biotin that catalyses acetyl-CoA carboxylation to malonyl-CoA, the rate-limiting enzyme for malonyl-CoA fatty acid synthesis. ACC1 is regulated by phosphorylation/dephosphorylation of serine residues [32]. The results of this study show that the expression of APOH plays a key role in regulating AKT/AMPK phosphorylation in myoblasts. AMPK is an upstream cytokine capable of activating apoptotic pathways. Phosphorylation of AMPK has been reported to have inhibitory effects on ACC1 in humans and mice [33,34]. AKT is a central regulator of lipid metabolism and an upstream signaling molecule that adjusts the Akt/mTOR pathway to stimulate lipid accumulation [35]. Based on our experimental data, APOH regulates the expression of ACC1 by inactivating the AMPK pathway and activating the AKT pathway to regulate the synthesis of fatty acids.
There is evidence that activation of AMPK can inhibit the differentiation of adipocytes by blocking the transcription factor PPARG [36]. PPARG gene is involved in the process of adipocyte differentiation, which may be due to the activation of AKT[37]. PPARG is the primary regulator of adipogenesis. Our data show that overexpression of APOH reduces the expression of PPARG and ACSL1, and knockdown of APOH increases the expression of PPARG and ACSL1. APOH may reduce the β-oxidation of fatty acids through the PPARG pathway to cause the accumulation of TG in CS2s. These pathways are the leading promoters of TG and CHOL accumulation. A similar study showed that high expression of ACSL1 in human liver cells reduces the β-oxidation of fatty acids through the PPARG pathway, thereby increasing TG [38]. The overexpression of ACSL1 in cardiomyocytes will increase the content of TGs in cells, which suggests that ACSL1 gene is involved in the accumulation of TGs in cardiomyocytes[39]. These results are contrary to the results of this experiment, which may be due to the different cell types selected, and different cell samples have large differences in the process of TGs metabolism. The above research results show that APOH inhibits the expression of PPARG by regulating the AMPK/AKT pathway. As a transcription factor, PPARG can participate in controlling TG levels and free fatty acid formation by regulating the expression of ACSL1 [38,40].
LPL is the main enzyme that converts TG into free fatty acids and it has important functions [16]. It is well known that PPARG is a downstream signal molecule of AKT and a transcription factor involved in regulating lipid metabolism [41-43]. After the mouse very-low-density lipoprotein receptor (VLDL) receptor gene was knocked out, the expression of LPL was significantly reduced, leading to a significant increase in plasma TG content after meals, which indicated that LPL is involved in physiological process of TG clearance in mice [44]. When we regulate the expression of APOH gene in myoblasts, we found that APOH inhibits the expression of LPL through the AKT/PPARG pathway, reduces the clearance of TG in myoblasts, and causes the accumulation of TG in cells.
ELOVL6 is a particulate enzyme involved in the elongation of long-chain saturated and unsaturated fatty acids [45]. Studies have shown that ELOVL6 can catalyse lipid synthesis in bovine mammary epithelial cells [46]. ELOVL6 has a central role in obesity-induced insulin resistance by altering fatty acid composition and is a major target of hepatic sterol regulatory element-binding proteins [47]. In addition, the long-chain fatty acid metabolism driven by ELOVL6 is regulated by AMPK signal transduction [48]. This study showed that overexpression of APOH increased the expression of ELOVL6, while knockdown of APOH had the opposite result. This result indicates that APOH may stimulate the expression of ELOVL6 by inhibiting the activation of APMK, thereby promoting the synthesis of long-chain fatty acids in CS2s.
CONCLUSION
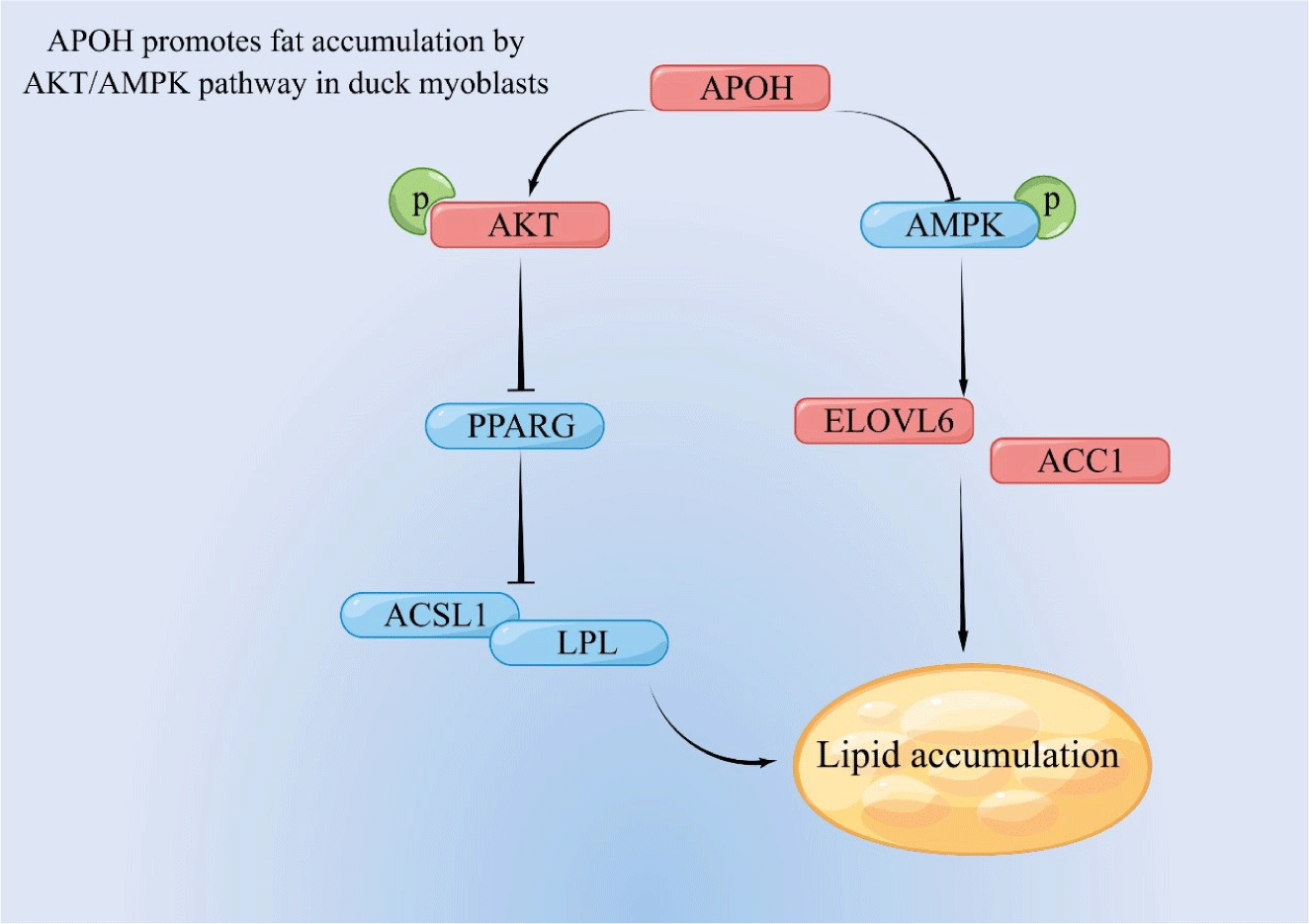
In summary, our results show that APOH inhibits the expression of PPARG and activating AKT, thereby inhibiting fatty acid oxidation, and at the same time, by inhibiting AMPK, it enhances the expression of ACC1 and ELOVL6 and promotes lipid synthesis (Fig. 9).