
INTRODUCTION
Enclosed housing with mechanical ventilation systems has recently become increasingly common in pig farming [1]. From such buildings housing animals at high densities, particulate matter (PM), which is a major air pollutant, is emitted and adversely affects human and animal respiratory health [2,3]. PM from pig houses comprises feces, feedstuff, hair, bedding particles, and animal skin, where feces and feeds occur as smaller particles than biological structures such as animal skin and hair [4], and feedstuff accounts for a considerable proportion in PM [5]. Airborne PM can lead to increased prevalence of respiratory diseases such as asthma [6] and chronic obstructive pulmonary disease [7] in humans. Moreover, airborne PM negatively affects respiratory health of pigs [8], and nursery pigs are more susceptible to respiratory disease than finishing pigs [9]. Tang et al. [10] suggested that swine respiratory diseases occur because of oxidative stress and inflammatory responses induced by PM.
Microbes contained in PM leaking from pig houses can spread by airborne transmission; however, culture-based methods used to investigate airborne microbes such as Escherichia coli in pig houses [9] do not suffice to resolve taxonomic diversity of such microbiomes owing to limitation of culture media [11]. Culture-independent methods such as next-generation sequencing can help identify various microbiota [11], and this approach was previously used to resolve the composition of airborne microbes in PM emitted from pig houses [12–15]. The most dominant bacterial phylum in pig houses is Firmicutes [12,13,15]; however, Aerococcus viridans, Bacillus cereus, Serratia marcescens, Vagococcus fluvialis, Clostridium spp., Enterococcus spp., Staphylococcus spp., and Streptococcus spp., which are potential airborne pathogens, have also been traced [16].
The community structure of airborne microbes in PM of pig houses may depend on factors such as environment, source, season, and air pollution levels [17]. Swine feces are the main component of PM [4], and they contain various microbes that are affected by diet, host genetics and age, and environment [18]. Therefore, identification of the microbiome composition in PM is of particular interest for improving pig productivity and health of pig farmers in South Korea. The objective of our study was to investigate microbiomes in PM emitted from pig houses in South Korea and to compare the differences in PM microbiomes between samples collected inside and outside of pig houses.
MATERIALS AND METHODS
Samples were collected in a pig farm housing approximately 9,000 pigs and located in Jangseong-gun, Jeollanam-do Province, Korea. In summer 2020, PM samples were collected at exhaust fans of a 1,000-head pig finishing building and an enclosed 390-head pig nursery section selected from eight enclosed sections in a nursery building (Fig. 1A). Outdoor PM concentrations were measured in an open space on a hill where the buildings’ ventilation systems were assumed to not directly affect air PM concentrations (Fig. 1B).

Three PM indices, PM2.5 (PM < 2.5 µm diameter), PM10 (PM < 10 µm diameter), and total suspended particles (TSP; PM smaller than approximately 50–100 µm diameter), were recorded simultaneously and gravimetrically using a cassette and two impactor samplers (PEM, SKC, Blandford Forum, UK) connected to portable air pumps (AirCheck, SKC). The air pumps provided a flow rate of 2 L/min for TSP and 4 L/min for PM2.5 and PM10.
Filters were weighed in a dehumidified chamber (30 ± 5% relative humidity) before and after sampling using a precise balance (BM-22, A&D, Tokyo, Japan). All filters were stabilized for 24 hours in the chamber and were then weighed three times. The weight increment after sampling was considered to represent the amount of PM, and PM concentration was calculated as the mass of collected PM divided by the air volume that had passed through the filter.
A set of instruments for measuring the three PM indices was installed within protective housing in front of exhaust fans and at the outdoor sampling site. Measurement of the three PM indices was carried out for 6 h (from 10.00 a.m. to 16.00 p.m.) on three different days in summer 2020. The exhaust fans were located on the side walls of the nursery building, whereas the exhaust fans of the pig finishing building were placed on the end wall in summer.
Using sterilized scissors, the filters capturing PM2.5, PM10, and TSP were cut in small pieces, which were then pooled based on the following three types: 1) three pooled PM samples collected on three different days (n = 3) from inside the nursery pig house (I-NPH), 2) three pooled PM samples collected on three different days (n = 3) from inside the finishing pig house (I-FPH), and 3) three pooled PM samples collected on three different days (n = 3) from outside of the pig houses (O-PH). After the filter pieces were transferred to bead tubes, metagenomic DNA was extracted using the bead-beating plus column (RBB+C) method [19].
Bacterial 16S rRNA gene amplicons including the V3-V4 hypervariable region were produced using primers 341F (5′-CCTACGGGNGGCWGCAG-3´) and 805R (5′-GAC TACHVGGGTATCTAATCC-3´) and were then subjected to high-throughput sequencing on an Illumina MiSeq platform (Illumina, San Diego, CA, USA) as described previously [20]. The resulting sequences were assembled using FLASH software [21], and microbiome analysis was conducted using the QIIME 1.9.1 software package [22] as described previously [23,24].
The proportion of total reads per taxon was log-transformed to produce a normal distribution. Log-transformed proportion values and DNA concentrations were compared among the three PM sample types (I-NPH, I-FPH, and O-PH) using an analysis of variance followed by Duncan’s multiple range test using XLSTAT statistical software version 2019.4.2 (Addinsoft, New York, NY, USA). Statistical significance is reported at p < 0.05. Spearman’s rank correlation was conducted to analyze correlations among PM concentrations, DNA concentrations, and major taxa using XLSTAT statistical software.
RESULTS
In total, 351,016 sequences were produced from the nine PM samples. Phyla or genera with an abundance of > 0.2% of all sequences, on average, were considered “major taxa” and were subjected to statistical analysis. Firmicutes was the dominant phylum accounting for 64.8%–97.5% of the sequences in individual samples, followed by Proteobacteria (1.4%–21.8%) and Bacteroidetes (0.3%–13.7%) (Fig. 2). The proportions of these three major phyla did not differ significantly (p > 0.05) among the three PM types. The remaining minor phyla accounted for < 0.1% of all sequences, on average, and included Spirochaetes, Deinococcus-Thermus, Candidatus Melainabacteria, Chloroflexi, Fusobacteria, Verrucomicrobia, Tenericutes, Acidobacteria, Gemmatimonadetes, Chlamydiae, Elusimicrobia, Fibrobacteres, and Planctomycetes.
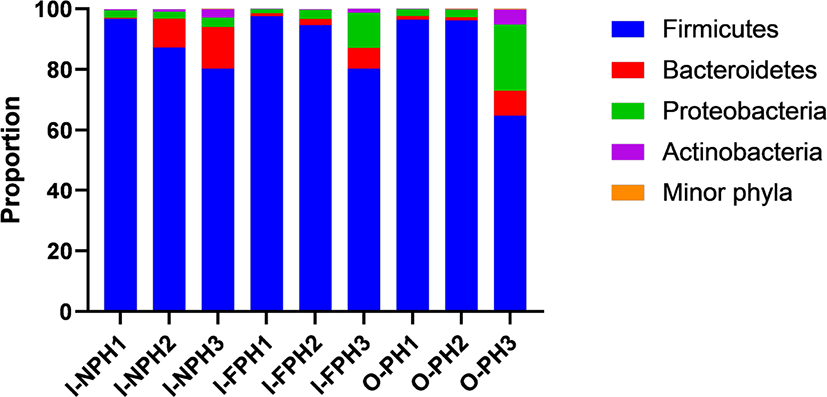
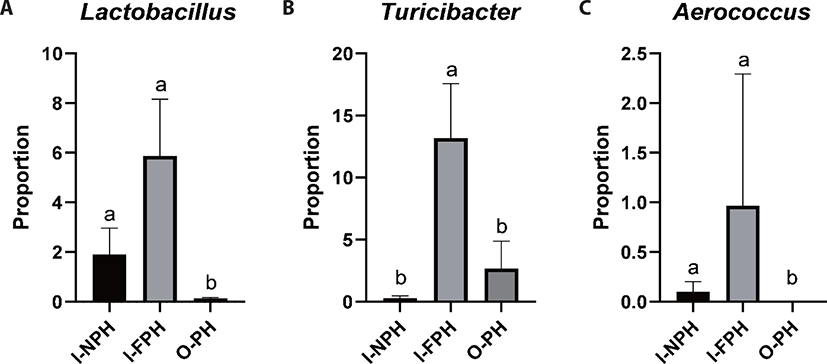
At genus level, Clostridium was predominant and accounted for 28.1% of sequences, on average, across the nine PM samples. The second dominant genus was Bacillus at 25.2%, on average, followed by Terrisporobacter (9.0%), Turicibacter (5.4%), Lactobacillus (2.6%), Prevotella (2.5%), Staphylococcus (2.2%), Curvibacter (2.2%), Weissella (1.6%), Sediminibacterium (1.2%), Roseburia (1.1%), and Blautia (1.0%). Genera accounting for < 1.0% of all sequences, on average, included Eubacterium (0.9%), Bradyrhizobium (0.8%), Pediococcus (0.7%), Faecalibacterium (0.6%), Mediterraneibacter (0.5%), Streptococcus (0.5%), Gemmiger (0.5%), Ruminococcus (0.4%), Hungateiclostridium (0.4%), Corynebacterium (0.4%), Sphingomonas (0.4%), Aerococcus (0.4%), Oscillibacter (0.4%), Barnesiella (0.4%), Pelomonas (0.3%), Flintibacter (0.3%), Phascolarctobacterium (0.3%), Holdemanella (0.3%), and Dorea (0.3%). Among the major genera, the proportion of Lactobacillus was significantly larger (p < 0.05) in the I-NPH and I-FPH samples than in the O-PH samples (Fig. 3A). The proportion of Turicibacter was significantly larger in the I-FPH samples than in the other two PM types, while that of Aerococcus was significantly larger (p < 0.05) in the I-NPH and I-FPH samples than in the O-PH samples (Figs. 3B and C).
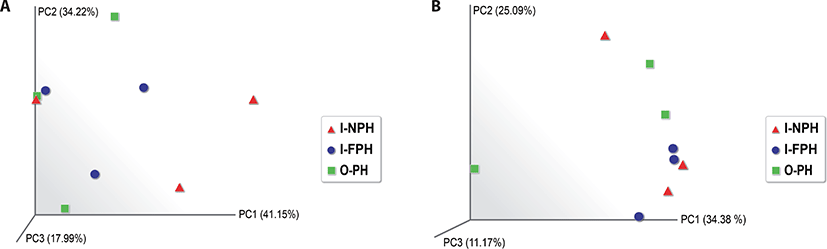
Alpha diversity analysis showed that the observed operational taxonomic unit (OTU), Chao1, Shannon, and Inverse Simpson indices did not differ significantly (p > 0.05) among the three PM types (Table 1). A beta diversity principal coordinate analysis (PCoA) based on weighted and unweighted UniFrac distances revealed that individual microbiomes of the nine PM samples were not distinct, indicating that the compositions of PM microbiomes were similar (Fig. 4).
| Particulate matter group1) | Observed OTUs | Chao1 | Shannon | Inverse Simpson |
|---|---|---|---|---|
| I-NPH (n = 3) | 245.33a | 253.28a | 4.00a | 0.75a |
| I-FPH (n = 3) | 268.00a | 300.54a | 3.49a | 0.76a |
| O-PH (n = 3) | 224.67a | 233.33a | 2.94a | 0.62a |
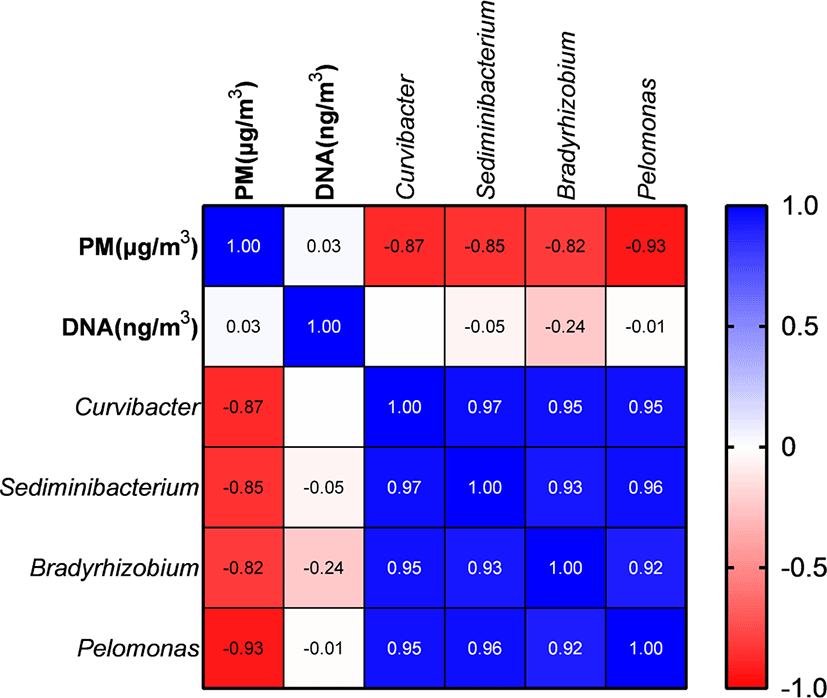
PM and metagenomic DNA concentrations did not differ significantly (p > 0.05) among the three PM types (Table 2). PM concentration did not show a significant correlation with DNA concentrations; however, it was significantly negatively correlated with the proportions of Curvibacter, Sediminibacterium, Bradyrhizobium, and Pelomonas (Fig. 5). The remaining major genera includingpathogens did not show a significant correlation with PM concentration.
| Variable | I-NPH (n = 3) | I-FPH (n = 3) | O-PH (n = 3) |
|---|---|---|---|
| Particulate matter (µg/m3)2) | 892.6 ± 200.1a | 1,282.2 ± 509.9a | 601.4 ± 239.8a |
| Metagenomic DNA (ng/m3)3) | 531.3 ± 209.0a | 481.3 ± 242.5a | 280.3 ± 140.0a |
DISCUSSION
Pig farming at high animal densities can lead to considerable emission of PM originating from feces, feedstuff, skin, and hair [15]. Potential pathogens in such PM can cause health problems among farmers and neighboring residents [25]. Swine breeds and diets vary among farms in different countries, and factors such as diet, host genetics and age, and environment can affect microbiomes in pig feces [18]. To the best of our knowledge, the current study is the first to assess the composition of microbiomes in PM inside and outside of pig houses in the Korea, and it may provide useful information to reduce potential risks associated with PM leaking from pig houses.
Previous studies reported that Firmicutes is the predominant phylum in PM from pig house, regardless of season [2,15,26], and our results are in line with these findings. As Firmicutes is the most abundant phylum in the pig gut microbiome during all growth stages [18], its high abundance in PM is likely due to bacteria originating from feces. Moreover, other major genera identified in the present study seemed to originate from feces. Clostridium was the predominant genus in PM, which may have various functions: Wang et al. [18] proposed that butyrate-producing Clostridium butyricum contributes to gut health, and its abundance is positively correlated with body weight. Clostridium herbivorans can degrade cellulose in enrichment cultures with swine feces [27], while some Clostridium spp. can ferment amino acids and produce ammonia [28]. As Clostridium spp. produce odorous products such as volatile fatty acids and ammonia, Clostridium has been considered one of major contributors to odor from swine farms [28]. Terrisporobacter was also identified as one of the dominant genera in the gut of pigs [29]. Bacillus spp. are typically used as feed additives and are abundant in pig feces [30], which may explain why it was the second most abundant genus in PM in the present study. Turicibacter contributes to increasing body weight and improving immune functions in pigs [18], while Lactobacillus spp. are commonly used as probiotics [31] which can degrade mycotoxins derived from contaminated feedstuff [32]. Lactic acid-producing Weissella has also been found in pig feces [33]. Prevotella, Roseburia, and Blautia produce short-chain fatty acids as fermentation products in the intestine of pigs [34]. Thus, most of the dominant microbes in PM from pig farms seem to originate predominantly from feces because of larger bacterial biomass in feces than in feedstuff, skin, and hair. Maintenance of pig gut health is thus important to reduce the prevalence of pathogens in PM emitted from pig farms.
Some genera in PM from pig houses were pathogens. Although most Clostridium spp. contribute to gut health and help increase body weight of pigs, some Clostridium spp. are potential pathogens [35]. Staphylococcus is the predominant genus on the skin of Korean people [36], and it is also common in pig houses and is part of the pig skin microbiome [37,38]. However, Staphylococcushyicus is associated with pig skin disease [37], and Staphylococcus aureus may cause skin or respiratory infections in humans and can be transmitted from pigs to humans via bioaerosols [39]. Although the assumed main function of Streptococcus spp. in the pig gut is to promote animal growth [18], Streptococcus suis is an important zoonotic pathogen in pigs and is ubiquitous in most countries [40]. The genus Escherichia including the pathogenic Escherichiacoli occurred at only 0.01% of all sequences in the present study. In addition, biosafety is important to prevent spreading of potential pathogens originating from sources other than swine feces.
Among the major genera, Clostridium and Bacillus spp. are typically predominant, not only in pig manure [41,42] but also in soil [43–45]. Therefore, pig manure and soil sources outside pig houses may contribute to the high abundance of Clostridium and Bacillus spp. in PM outside a pig house, whereas pig feces are presumably the source of bacteria of these two genera that occur at high abundances in PM inside the pig house. It seems that microbe-containing PM originating from inside the pig house leaks to the outside and then mixes with microbe-containing PM originating from outside the pig house. The mixed microbe-containing PM is thus presumed to have re-entered the buildings.
Lactobacillus, which plays an important role in maintaining gut health, was more abundant inside the pig house than outside. This is probably because Lactobacillus is predominant in feces of piglets and finishing pigs but not in sources outside the pig houses such as pig manure and soil. The abundance of Turicibacter increases with increasing body weight; thus, this genus is more abundant in finishing pig houses than in piglet houses [18]. Aerococcus spp. are pathogenic and have been isolated from clinical specimens of pigs [46,47]. In the present study, highly abundant Aerococcus inside the pig house seemed to be a result of infections in pigs. A previous study also reported that Aerococcus is highly abundant in PM from pig houses [16]. Farm workers may potentially be exposed to this pathogen contained in PM inside of pig houses. Therefore, reducing potential pathogens in pigs may help mitigate health problems in farmers and neighboring residents.
Alpha diversity indices and the beta diversity PCoA demonstrated that microbiomes were similar inside and outside of the pig houses. It seemed that most microbe-containing PM from inside pig houses leaked to the outside and then re-entered the buildings. Therefore, an increase in potential pathogen abundance and PM containing feces may cause health problems in farmers and neighboring residents. Maintaining pig gut health may help reduce the prevalence of fecal pathogens and mitigate potential risks associated with PM from pig houses. As described above, microbes from pig manure outside pig houses may mix with PM and then re-enter the buildings, which may also explain the similarity of the respective microbiomes.
PM concentrations were not correlated with the abundance of major genera, including pathogens in the pig gut. A previous study suggested a positive correlation between pathogen abundance and PM concentration [48], which was not confirmed by the results of the current study. The abundance of pathogens seems to be influenced by pig gut health rather than by the amount of feces; thus, maintaining gut health may be an important factor to help reduce the abundance of pathogens in PM. Abundances of Curvibacter, Sediminibacterium, Bradyrhizobium, and Pelomonas were negatively correlated with PM concentrations; however, these genera typically originate from soil or water [49–52], and even though PM concentrations increase, abundances of microbes originating from soil and water in PM may remain similar, whereas abundances of microbes from pig feces may be variable depending on pig gut health.
CONCLUSION
Our results suggest that pig feces are the main source of the PM from pig houses and of most of the dominant microbes in PM. Pathogen abundance was not correlated with PM concentrations, and pig gut health seems to affect the prevalence of pathogens. The overall composition of PM microbiomes was similar inside and outside of pig houses. However, among the two predominant genera Clostridium and Bacillus, some species seemed to originate from feces deposited inside the pig houses, whereas other species seemed to originate from pig manure and soil sources outside the pig house. It seems that microbes in PM inside pig houses leak to the outside and then mix with microbes in PM outside, after which they re-enter the buildings. Maintenance of pig gut health, as well as biosafety inside and outside of pig houses, may help reduce potential risks associated with pathogens in PM inside and outside of pig houses.