
INTRODUCTION
Methane (CH4) emissions from dairy cattle pose a significant environmental challenge, notably contributing to the global greenhouse gas footprint of agricultural activities. Therefore, effectively mitigating CH4 in dairy production is crucial to meet global environmental targets, such as those outlined in the Global Methane Pledge [1]. In this context, dietary interventions have recently garnered increasing attention as a promising approach to reduce enteric CH4 emissions from ruminants.
Among these interventions, the use of essential oil-based additives, such as Agolin® Ruminant (AR, Agolin SA), has gained attention for its potential to modify rumen fermentation and decrease CH4 output. Agolin® Ruminant is an essential oil (EO) product containing a blend of coriander seed oil, eugenol, geranyl acetate, and geraniol [2] geared towards reducing CH4 emissions in cattle and some agencies have issued carbon credits to producers utilizing the product. This product is also available in an organic certified carrier for use in organic farms. The EOs in this product presumably disrupt the phospholipid membrane of archaea, leading to a decrease in CH4 production and an increase in the cow’s nutrient utilization efficiency [3]. Previous studies have employed both in vitro and in vivo experiments, revealing the additive’s capacity to alter rumen fermentation patterns and reduce CH4 production [2,4]. For example, studies have highlighted the efficacy of EO in lowering CH4 emissions in dairy cattle [5], coinciding with improvements in milk production and components in response to EO supplementation [6]. However, the effects of EO on the rumen microbiome and their association with CH4 mitigation remain largely unexplored.
One of the main concerns regarding the administration of AR (as well as other EO-based products) is whether it also disrupts the rumen fibrolytics when fed to cattle, as very few studies have characterized the changes in the microbiome in response to Agolin® Ruminant [7]. In a recent study, AR fed in dual-flow continuous culture (DFCC) decreased CH4 production by 10% in less than two weeks of adaptation [8] but did not decrease fiber digestibility nor did it significantly alter volatile fatty acid production, prompting researchers to investigate which rumen microbes might be impacted by AR to both explain the mechanisms underlying decreased CH4 emission and gauge potential drawbacks.
To address this gap, our study employed samples from the previous DFCC study coupled with advanced microbiome analysis to investigate the effects of EO on rumen microbial populations and CH4 emissions in dairy cattle. This approach allows for a controlled simulation of the rumen environment, providing detailed insights into the microbial dynamics within the rumen and their relation to CH4 production [9].
This study aimed to determine the microbial factors associated with CH4 inhibition by EO using a DFCC system. We hypothesized that EO either decreased the protozoal population or induced shifts in the archaeal or bacterial abundance to decrease CH4 production. This study contributes to the evolving field of enteric CH4 mitigation in dairy cattle, offering valuable insights for the development of sustainable and effective dietary strategies in the dairy industry.
MATERIALS AND METHODS
The DFCC system utilized for this study implements updated approaches previously characterized [9]. Effluent samples used for DNA extraction and protozoal enumeration came from a previous study [6] evaluating the efficacy of several organic-certified CH4 inhibitors. Briefly, DFCC (n = 4) were inoculated from two Jersey cows housed at the Waterman Dairy Farm (The Ohio State University, Columbus, OH) under care according to the Institutional Animal Care and Use Committee protocol #2013A00000073. These cows were fed a lactating diet common to the herd at the time [10] including 39% corn silage, 9.9% wet brewer’s grains, 7.7% soybean meal, 4.6% bypass soy, 0.9% bypass fat, 0.9% molasses, and 3.4% vitamin mineral premix (without monensin or other feed additives). A Latin square treatment arrangement was applied to evaluate four treatments which are detailed previously. Of these, only two were considered for the current study due to a lack of efficacy previously reported for the other two treatments. The control (CON) diet was fed twice daily a total of 80 g/d dry matter (DM) diet (60:40 concentrate:orchardgrass pellet mix, 17.1% crude protein [CP], 33.0% neutral detergent fiber [NDF], 0.1% acid detergent fiber [ADF], and 27.1% starch; Table 1) and one fed CON diet with 3 mg/d dose of supplemental organically-certified EO (Agolin® Naturu, Agolin SA). Fermenter effluent samples (d8-11) were subsampled and stored at −80°C for metagenomic DNA extraction. Additionally, fermenter samples (d5) and effluent samples (d8-11) were fixed in formalin and stored for enumeration based on the procedure outlined in Dehority, 1984 [11]. The outflow of cells in effluent was contrasted to the fermenter contents populations to estimate generation time for protozoa, and protozoa were divided into the following types: Charonina (based on previous reports of high enrichment in DFCC [12]), Isotrichidae, Diplodinium, and other entodinia – either small (< 100 µm in length) or large (> 100 µm in length).
| Nutrient | Diet composition |
|---|---|
| Dry matter (g/d) | 80.0 |
| Crude protein | 17.1% |
| Starch | 27.1% |
| Water soluble carbohydrates | 8.4% |
| Neutral detergent fiber | 33.0% |
| Acid detergent fiber | 20.1% |
| Fat | 2.2% |
| Ash | 9.2% |
Metagenomic DNA from the effluent samples was extracted using the repeated bead beating plus column method [13] and purified with Qiagen mini-stool kits from Thermo Fisher Scientific. The researchers conducting the extractions were blinded to treatment during the DNA extraction and subsequent taxonomy classification. The quality and quantity of DNA were assessed using a NanoDrop ND-2000 spectrophotometer (Thermo Scientific, NanoDrop Technologies) and further evaluated through 1% agarose gel electrophoresis. Amplicon libraries, targeting the V4 hypervariable region of the 16S rRNA gene were generated using the 515F and 806R universal primer pair [14] and each library was uniquely barcoded for multiplexing at The Ohio State University’s Molecular and Cellular Imaging Center (Wooster, OH). The libraries were then pooled and sequenced using an Illumina MiSeq sequencer (2 × 300 bp paired-end sequencing). Quality control measures, including denoising, merging, and chimera removal, were performed using QIIME2 version 2022.2 [15], following an approach similar to that described previously [16].
In this analysis, the final number of quality amplicon sequencing variants (ASVs) was 3,662,173 (3,653,369 bacterial ASVs and 8,804 archaeal ASVs) and they were classified taxonomically based on a 99% similarity . This classification was conducted using the weighted Silva 16S pre-trained classifier (NR 138 version; [17–19]) to enhance classification accuracy. Only phyla, families, and genera with a relative ASV abundance ≥ –0.5% in at least one treatment were included in the analysis. ASV BIOM tables for archaea and bacteria were separated prior to downstream analysis. Alpha diversity indices such as richness, Chao1, Shannon’s index, Pielou’s evenness, Good’s coverage, and Faith’s phylogenetic diversity were derived from the average rarefied ASV table (repeated 100 times, referenced in [20]). The microbial metabolic functions were predicted using PICRUSt2 [21] utilizing 16S ASVs. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were reconstructed from these predictions, representing microbial metabolic functions based on KEGG ortholog profiles derived from PICRUSt2.
Statistical assessment of the microbial relative abundance data was carried out using MaAsLin2 [22] to examine the impact of EO. This analysis involved centered log-ratio normalization and a linear model without data transformation. The statistical model encompassed the fixed effects of both the treatment and fermenter, along with the random effect of the period. The mixed model was implemented using either MaAsLin2 or SAS 9.4 (SAS Institute), particularly for analyzing effluent protozoal counts. Alpha diversity metrics and effluent protozoal counts were examined using the SAS mixed model. A significance threshold of Q < 0.05 (Benjamini-Hochberg FDR-corrected P-values) was set for MaAsLin2 analyses, and a p < 0.05 was used for SAS analyses. Bray-Curtis and Jaccard distance matrices were compared to evaluate the overall microbial community differences resulting from EO. This was done using the adonis2 function within the ‘vegan’ package (version 2.5–7) of R [23]. The same statistical models were applied to conduct these comparisons. Additionally, PCA results were graphically represented through plots created using the ‘ggfortify’ package in R (version 3.5.3) [24].
RESULTS
Protozoal populations were primarily entodiniomorphids (74%, Table 2) – mostly shorter in length than 100 µm – while another 16% of the protozoal population was Charonina spp. Both protozoal populations within fermenters and daily flow of protozoa in effluent were unchanged by treatment, as was generation time (p > 0.10). In the control treatment, no members of Isotrichidae were detected in effluent samples despite being identified in fermenter populations.
Fig. 1 provides a comprehensive overview of the primary bacterial communities identified in the effluent samples at the phylum level. Our analyses revealed five major phyla – each representing more than 0.5% of the average relative abundance in either the control or treatment groups – accounting for 98.4% of the relative abundance of the detected ASVs. These phyla include Bacteroidota (45.6%), Firmicutes (27.6%), Proteobacteria (19.7%), Spirochaetota (3.5%), and Patescibacteria (1.9%) (Fig. 1A). At the genus level, 34 major bacterial genera accounted for 90.3% of the total bacterial population, as shown in Fig. 1B.
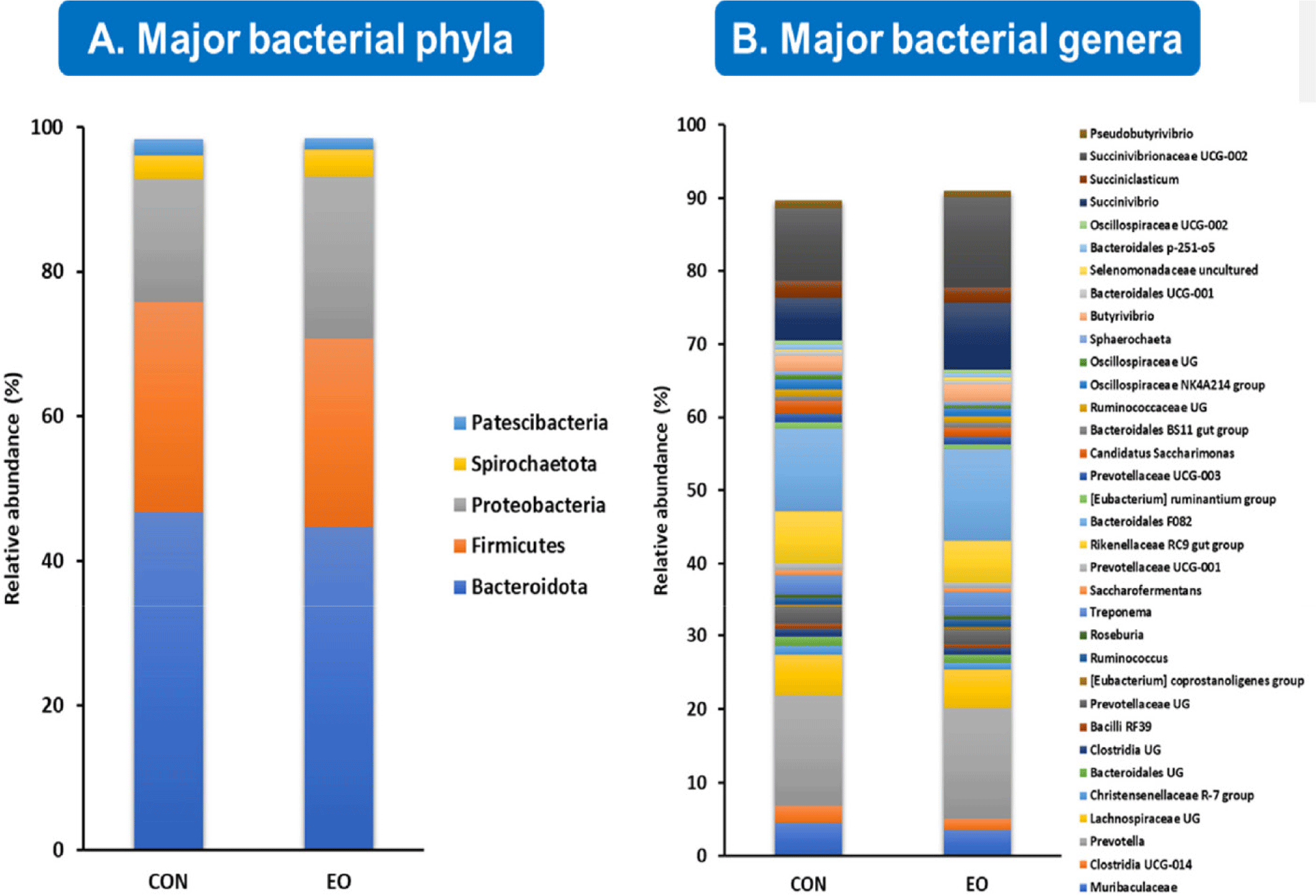
In terms of archaeal communities, all ASVs were classified into two families: Methanobacteriaceae (97.2%) and Methanomethylophilaceae (2.8%), with only three genera detected (Methanobrevibacter, Methanosphaera, and uncultured genus within the Methanomethylophilaceae family). Notably, all major bacterial and archaeal taxa were identified in both the control and EO treated groups.
This study also investigated the impact of EO on the diversity of the prokaryotic microbiome, revealing a significant reduction in all alpha-diversity indices – including species richness, evenness, phylogenetic diversity, and comprehensive indices (Shannon’s index and Simpson’s index) – across both the bacteriota and archaeota (Table 3). Despite these changes, the overall composition of the prokaryotic microbiome, as assessed by Bray-Curtis and Jaccard distance matrices, remained unaffected by EO treatment (p > 0.1; Fig. 2).
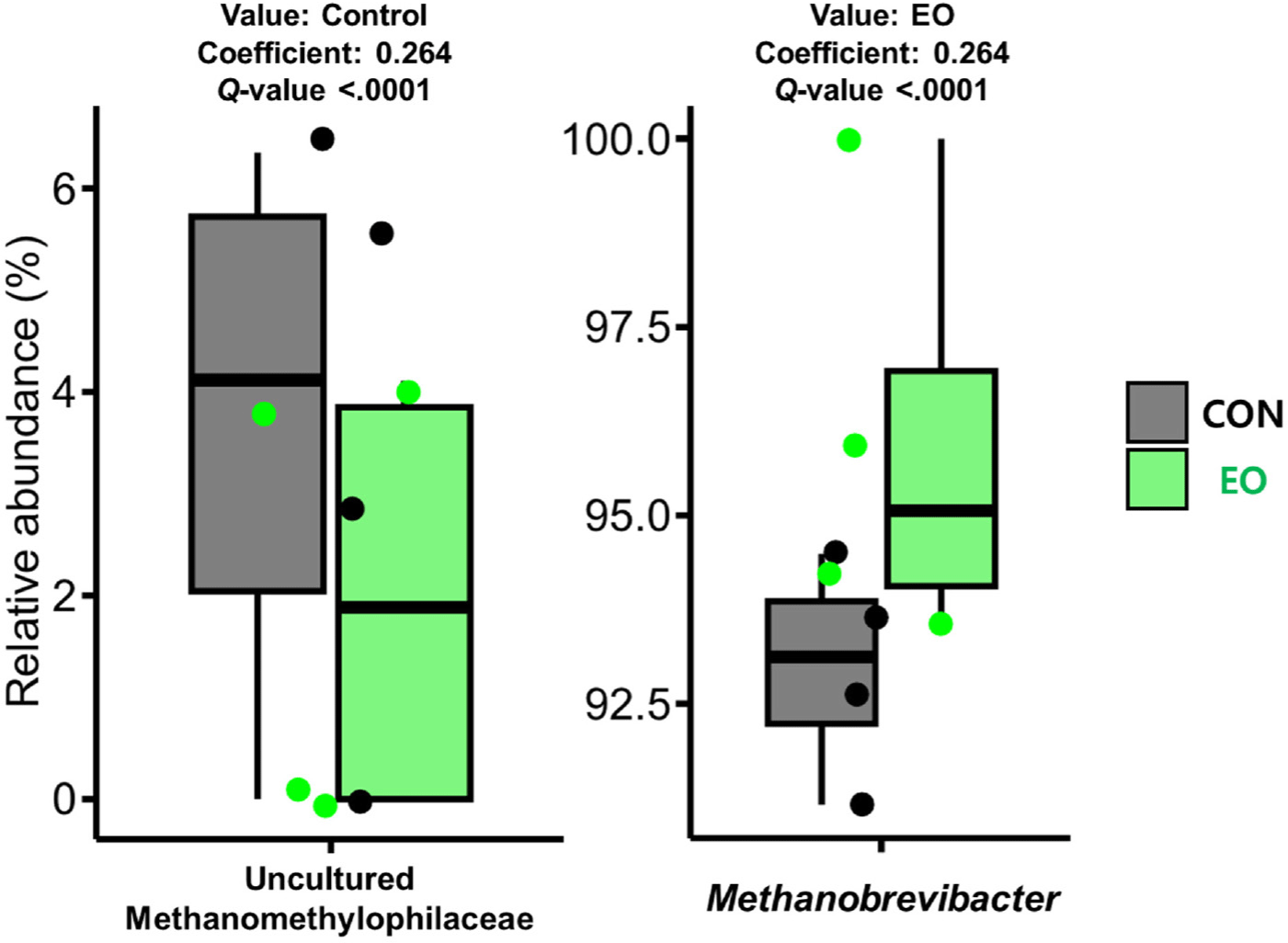
Comparative analysis between the control and EO-treated groups at the phylum level revealed differential abundances in Proteobacteria, Spirochaetota, and Patescibacteria (Table 4). At the genus level, eight bacterial genera (Clostridia UCG-014, Christensenellaceae R-7 group, unclassified genus within Bacteroidales, Bacilli RF39, Prevotellaceae UCG-001, Rikenellaceae RC9 gut group, and Candidatus Saccharimonas) exhibited a positive association with the control group, whereas four genera (Ruminococcus, Treponema, Butyrivibrio, and Succinivibrionaceae UCG-002) were more closely associated with the EO treatment (Table 4). Moreover, among the archaeal genera, Methanobrevibacter and an uncultured Methanomethylophilaceae genus exhibited positive and negative associations with EO treatment, respectively (Fig. 3).
An examination of the major KEGG pathways predicted from the bacterial communities revealed differential abundances in 26 pathways between the control and treatment groups as shown in Table 5. Similarly, for the archaeal microbiome, differential abundances were observed in seven and eight major KEGG pathways between the control and EO treatment groups (Table 6).
DISCUSSION
Prior research indicated that EO did not significantly alter fermentation characteristics, such as VFA profiles, various nutrient digestibility estimates, and ammonia concentration in effluent samples from this DFCC experiment but there was a 10% decrease in CH4 for this study [25], aligning with the findings of a meta-analysis of the effects of AR treatment [2]. Our findings also demonstrated that EO had no impact on protozoal populations, including the specific genera identified in the present study. This is particularly noteworthy, as protozoal inhibition has been suggested as a potential mechanism for EO-induced CH4 mitigation by targeting hydrogen producers and the protozoa-associated methanogen community [26–30]. However, previous studies on various EOs have not consistently demonstrated this effect [31], highlighting the need for further research to elucidate the potential mechanisms through which EOs influence CH4 production under different ruminal conditions. Further, DFCC systems modified for protozoal retention tend to harbor fewer protozoa – a drawback which has been documented in previous work [12] and the specific effect of EO on protozoa is recommended for further investigation in other in vitro models.
In the current study, analyses using Bray-Curtis and Jaccard matrices revealed that the overall archaeal and bacterial microbiome remained unaffected by EO treatment, aligning with results from previous studies on EO treatments both in vitro and in vivo [32–34]. Despite minor microbial shifts, the overall microbiome exhibited relative stability.
EO treatment decreased the alpha diversity of the archaeota and bacteriota, which was consistent with findings from an in vivo study on lactating dairy cows [7]. This reduction in prokaryotic diversity could be linked to more efficient feed utilization and decreased CH4 emissions [35]. Previous EO treatments have been reported to inhibit methanogens and decrease CH4 production [31,36–38], suggesting that the observed lower methanogenic diversity and abundance of methylotrophic methanogen might result from the direct inhibitory effects of EOs. However, further research, particularly involving AR treatment, is needed to validate these suggestions.
Carbohydrate-fermenting Clostridia such as Clostridia UCG-014 and Christensenellaceae R-7 group, which accounted for a significant portion of ruminal hydrogenase transcripts in a previous study [39], play a key role in hydrogen production. Therefore, reducing the abundance of these primary hydrogen producers could decrease CH4 production. The differential distribution of two butyrivibrios might affect fermentation characteristics, especially butyrate production, due to their similar phenotypic characteristics but distinct phylogenetic classification [40]. Propionate-producing bacteria such as Succinivibrionaceae UCG-002, commonly found in the CH4-inhibited conditions in the rumen of dairy cattle [41,42], were prevalent in the EO treatment, supporting previous observations of numerically increased propionate levels with EO treatment [25]. The abundance of Spirochaetes, specifically Treponema spp., was significantly higher in the EO treatment, and also exhibited greater abundance in EO-treated lactating dairy cows [7]. However, the metabolic versatility of Treponema spp. makes it difficult to pinpoint the exact reason for their increased abundance in response to EO treatment [43].
The inhibition of CH4 production could stimulate anabolic processes requiring metabolic hydrogen, such as fatty acid synthesis [44]. Although we did not observe significant changes in metabolic hydrogen concentrations previously for this study [24], shifts in microbial pathways related to these fermentation end-products are anticipated following EO treatment.
Among the KEGG pathways related to CH4 metabolism (ko00680), the control group exhibited significant differences in the relative abundance of pathways involved in the pentose phosphate pathway, glycine, serine and threonine metabolism, glyoxylate and dicarboxylate metabolism, and carbon fixation pathways in prokaryotes, whereas EO treatment stimulated pathways related to the metabolism of cofactors and vitamins including riboflavin metabolism and folate biosynthesis.
Acetogens require folate to produce acetate and ATP by reducing two molecules of carbon dioxide through the Wood-Ljungdahl pathway [45,46]. Therefore, although we did not detect significant changes in acetogen growth, our findings suggest that there was a shift in their growth patterns, especially under conditions that inhibited CH4 production. The stimulation of riboflavin metabolism, associated with the biosynthesis of coenzyme F420 [47], could potentially counteract EO-mediated methanogenesis inhibition. However, this function and other vitamin B complex metabolic functions were also more prevalent in the rumen microbiome of cows exhibiting high milk yield and milk protein content [48]. Additionally, archaeal metabolic pathways enriched in response to EO administration, such as the biosynthesis of secondary bile acids and ABC transporters, were found to be negatively correlated with CH4 emissions [49], suggesting that they play a key role in modulating the microenvironment and facilitating host-microbiome interactions [50–52].
Collectively, our findings suggest that the lack of substantial effects of EO on fermentation and digestion parameters measured parallel to the current study [24] might be due to the absence of a direct inhibitory effect of EO on methanogens. Offsetting shifts in the relative abundance of fiber-degrading bacteria and detailed methanogen communities deserve further investigation including predicted metabolic pathways impacted by population changes. It appears that EO moderates CH4 production primarily by modulating the ruminal prokaryotic microbiome.