
INTRODUCTION
Major histocompatibility complex (MHC) is a group of closely linked genes that forms a dynamic genetic component of the mammalian immune system. Bovine MHC isolation and characterization were determined long years ago [1–3]. MHC molecule is the primary component of adaptive immunity in bovines and the two classes of MHC genes (class I and class II) are the main gears of the MHC region; typically the most polymorphic regions of the gene in the majority of the mammalian genome [4]. MHC molecule is physically mapped on the bovine autosome 23 (BTA 23). The MHC of cattle is called bovine leukocyte antigen (BoLA), which is highly variable and helpful in the immune systems. The role of BoLA in cattle is to recognize pathogens via the immune systems and mainly used as a disease marker in cattle breeding programs [4,5].
BoLA-DRB3 locus is the most widely studied and polymorphic region in bovines. The allele diversity and polymorphisms of the DRB3 gene in cattle breeds have recently been reported by sequence-based typing [6]. According to the report available in immuno-polymorphism database (IPD-data base) 136 alleles are deposited so far; by which such impressive polymorphisms have resided on the β1 domains of the peptide-binding regions (PBR) [7,8]. Among the six exons of BoLA-DRB3 gene, exon 2 has previously been reported to be the most polymorphic and potentially affect many immunities, somatic cell count and mastitis incidence [9]. Moreover, recent studies reported that the BoLA DRB3 gene has been associated with susceptibility or resistance to mastitis [10,11].
Polymorphisms of BoLA-DRB3.2 in Korean cattle and Holstein cattle were characterized using a next generation sequencer [12] and aimed at the identification of molecular markers linked to bovine diseases and immunological traits by comparing other unique cattle breeds from developing countries like Bangladesh and Ethiopia.
Red Chittagong (RC) is one of the promising cattle varieties of Bangladesh. They are well adapted to traditional husbandry practices, subsistence on poor quality feeds and fodders. They have been breeding regularity and better resistance capabilities to withstand environmental stress and tropical diseases [13]. Yet, the BoLA DRB3.2 gene has not been studied in the RC cattle breed.
Ethiopia harbors diverse cattle populations adapted to a range of harsh environments. Sheko cattle are well adapted to humid environments of south-western Ethiopia characterized by high tsetse fly infestation [14]. In contrast, cattle populations such as Boran, Ogaden, and Afar have long been evolved under arid and semi-arid areas of eastern and southern Ethiopia where the risk of trypanosomiasis is relatively low. Despite some differences in their response to local diseases and parasites, to the best of our knowledge, no study has been made to characterize the MHC BoLA DRB3.2 gene in Bangladesh and Ethiopian cattle breeds which are critically important for future breeding programs. The aim of this study was, therefore, to characterize the genetic variability of DRB3.2 in the African and Asian zebu and taurine cattle breeds.
MATERIALS AND METHODS
A total of 100 animals comprising of four Ethiopian indigenous cattle (Boran, n = 13; Sheko, n = 20; Fogera, n = 16; Horro, n = 19), Korean Hanwoo cattle (n = 18) and Bangladesh Red Chittagong zebu (n = 14) were considered in this study (Table 1). These samples were randomly selected from multiple herds and 102 additional Holstein Freisian sequence samples were downloaded from the National Center for Biotechnology Information (NCBI), included in the analysis.
Nasal swabs were collected using an Animal Swabs Collector (BlueGene Life Science, Cheongju, Korea) and Performagene LIVESTOCK’s nasal swabs (DNA Genotek, Kanata, ON, Canada). DNA was isolated from nasal swabs following the manufacturer’s instructions.
The quality of the DNA and its concentration were quantified via NanoDrop1000 and electrophoresis in 0.8% agarose gels. Those DNA samples with good quality and quantity were considered for amplification and sequencing.
To amplify exon2 of the BoLA-DRB3 gene, we used primers reported in the previous study [15]. PCR technique amplified 284 bp product at the second exonic region. The reactions were carried out in a total volume of 30 μL containing genomic DNA (2 μL of 50 ng/μL), 21.1 μL distilled water, 10 × PCR buffer (3 μL), 10 mM dNTPs (0.6 μL), forward and reverse primers (1.5 μL of 10 pM for each), and 0.3 μL ofTaq DNA polymerase (Promega, San Diego, CA, USA).
Reactions were performed; at 35 cycles of 94°C for 30 s, annealing at 60 °C for 30 s, and 72°C for 40 s preceded by initial denaturation at 94°C for 10 min and a final extension at 72°C for 5 min. Finally, the PCR products were visualized by gel electrophoresis on 2% agarose gels with acetate EDTA (TAE) buffer followed by ethidium bromide staining. Sequencing was performed by Macrogen (Seoul, Korea).
We conducted PCR-SBT technique to evaluate the genetic variability of BoLA-DRB3.2 gene. The study was pioneered for characterization of the BoLA DRB3.2 region in the Ethiopian cattle breeds. Polymorphism of the BoLA-DRB3.2 region was investigated in four Ethiopian local breeds (Boran, Fogera, Horro, and Sheko) and Korean Hanwoo and Bangladesh Red Chittagong. The DNA sequences were edited using Bio-edit version 7.0.5.3 and aligned by clustalX2 software package [16]. DNA polymorphism was computed based on six parameters (number of polymorphic sites, the total number of mutations, the average number of nucleotide differences, number of haplotypes, haplotype diversity and nucleotide diversity) using ARLEQUIN software version 3.5.2.2 [17]. Also, the allele frequencies and number of alleles were estimated by the same software. The deviations from Hardy Weinberg equilibrium (HWE) were computed by F-statistics [18]. Moreover, observed and unbiased expected heterozygosity was calculated by ARLEQUIN and GENPOP4.0 software [19]. Pattern of sequence variability, synonymous and non-synonymous substitutions were computed by DnaSP version 6.12.03 software [20]. Nucleotide similarities of the newly identified alleles with the reference sequence (BoLA-DRB3:016:01) were generated by the sequence demarcation tool [21]. The average haplotype count per individual (AHC) was calculated by dividing the number of haplotypes by the number of animals considered [22].
Population differentiation due to the population genetic structure was also assessed from sequence data, population pairwise Wright’s FST [18] values were calculated by using ARLEQUIN software version 3.5.2.2 applying 1000 replication values [17]. The pairwise FST graph was displayed by Rcmd (console version of the R statistical package) installed on the computer integrated with ARLEQUIN (http://www.r-project.org/). Moreover, population genetic differentiations based on the BoLA-DRB3 gene of the breeds were evaluated by Ne’s genetic distance (GST) by DnaSP software. Molecular diversity index statistics, the number of transitions and numbers of transversions were computed by ARLEQUIN software version 3.5.2.2 [17].
Neutrality tests were performed by infinite site model using ARLEQUIN software. Tajima’s D and Fu’s Fs neutrality tests were estimated based on the mean number of pairwise differences between haplotypes and probability of observing an average number of nucleotides (number of alleles), respectively [23,24].
Phylogenetic analysis was carried out using the Maximum Likelihood method based on the Kimura 2-parameter model [25] using MEGAX 10.1 software [26] via implementing1000 bootstrap values [27]. Positions from DNA sequences containing gaps or missing data including identical sequences were excluded from the analysis.
Median-joining network (MJN) tree was constructed using the NETWORK software (version 10.0.0) [28]. To evaluate the median network, the nucleotide sequences were first converted into binary data, while identical sites were omitted from the analysis. Each split was programmed as a binary character, satisfying the values of 0 and 1. The haplotypes were denoted as a binary vector in this method. The median vectors were estimated for each triplet of vectors until the construction of the median network was achieved [28].
RESULTS
PCR-SBT analysis of BoLA-DRB3.2 resulted in the identification of 59 BoLA-DRB3.2 alleles of which 16 alleles (BoLA-DRB3*164:01, BoLA-DRB3*165:01, BoLA-DRB3*166:01, BoLA-DRB3*167:01, BoLA-DRB3*168:01, BoLA-DRB3*169:01, BoLA-DRB3*170:01, BoLA-DRB3*171:01, BoLA-DRB3*172:01, BoLA-DRB3*173:01, BoLA-DRB3*174:01, BoLA-DRB3*175:01, BoLA-DRB3*176:01, BoLA-DRB3*177:01, BoLA-DRB3*178:01 and BoLA-DRB3*179:01) were reported here for the first time. All sequences were deposited in the NCBI gene bank data base with accession numbers MT919537-MT919549 (Bangladesh), MT919550-MT919562 (Boran), MT919563-MT919578 (Fogera), MT919579-MT919596 (Hanwoo), MT919597-MT919615 (Horro), and MT919616-MT919637 (Sheko). Among 59 different alleles, 11 alleles in Bangladesh Red Chittagong, 12 in Boran, 13 in Fogera, 8 in Korean Hanwoo, 14 in Horro and 18 in Sheko were detected. Alleles were named temporarily based on the principles of IPD-MHC database.
The allele BoLA-DRB3*145:01 shared between the Bangladesh Red Chittagong and two Ethiopian breeds (Boran and Horro). The allele BoLA-DRB3*100:01 was recorded only in three Ethiopian breeds (Boran, Fogera, and Horro). The Korean Hanwoo breed shared BoLA-DRB3*021:01 allele with three Ethiopian breeds (Fogera, Horro, and Sheko). However, no alleles were shared between Korean Hanwoo and Bangladesh Red Chittagong. Moreover, BoLA-DRB3*002:01 allele had a higher frequency of 0.444 while the lowest frequency was 0.050 which was observed for many alleles (Table 2).
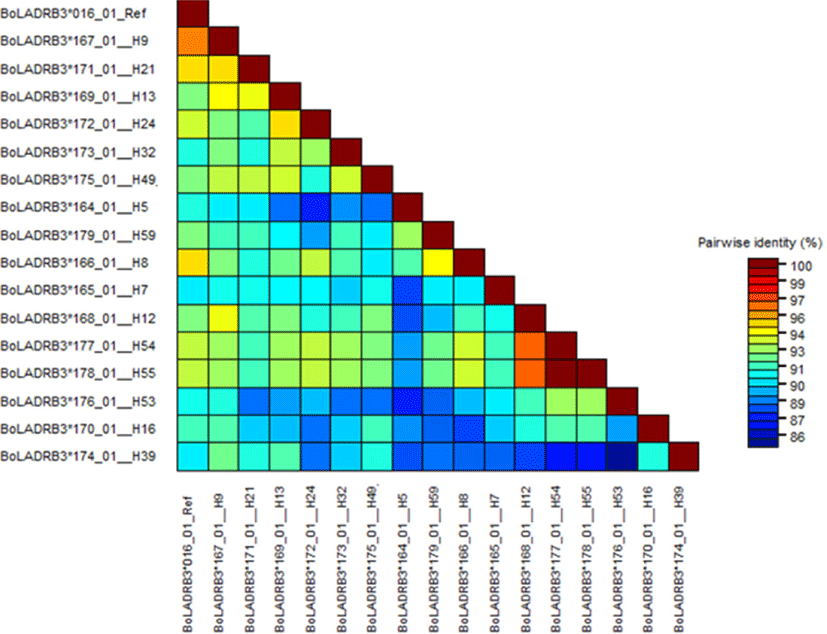
The nucleotide sequences of the newly identified alleles were compared with the reference sequence from the IPD-MHC data base (BoLA-DRB3*106: 01) and higher variability of the region was visualized (Fig. 1). The nucleotide identity of the new alleles identified from this study compared to the reference sequence from IPD-MHC data base showed 88%–100% similarity (Fig. 2).
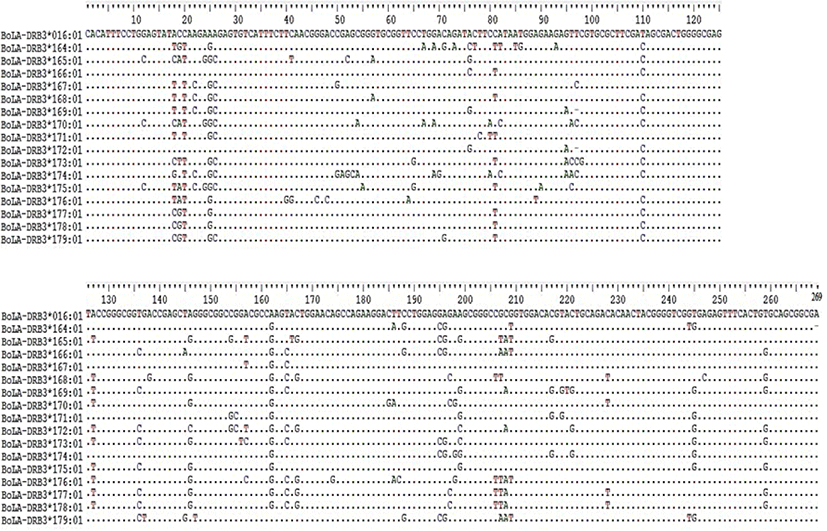
The number of polymorphic sites (S) which is the measure of usable loci that show more than one allele per locus was analyzed. The value of S was higher in Sheko (65) and lower in Hanwoo (47). Bangladesh Red Chittagong and Ethiopian Fogera had the same number of polymorphic sites (53). The nucleotide diversities (Π) were relatively high in Boran breeds (0.072) whereas in the Korean Hanwoo breed a relatively low number of Π (0.056) was observed. For all breeds, synonymous substitutions were less as compared to the non-synonymous substitutions (Table 3). Haplotypes were constructed for each breed and a total of 59 haplotypes were obtained. Of the six cattle breeds, the haplotype values ranged from 18 (Sheko) to 8 (Korean Hanwoo). The haplotype diversity (Hd) of the Ethiopian breeds and Bangladesh Red Chittagong were relatively higher as compared to the Korean Hanwoo. The haplotype diversity for Korean Hanwoo was 0.751 which was the lowest as compared to the rest of the breed. The average number of haplotype count per animal varied from 0.500 in Hanwoo to 0.846 in Ethiopian Boran. The AHC for Sheko was higher next to Boran (0.800). The average numbers of nucleotide differences (NPD) were highest in Boran (18.307) and lowest in Hanwoo (14.339). Moreover, the ratio of non-synonymous substitution (NSs) to synonymous substitution (SS) was highest in Sheko with an estimated value of 3.300 and lowest in Korean Hanwoo with an estimated value of 1.500 (Table 3).
To understand genetic diversity within a population, the mean number of alleles per locus (Na), the average expected (He) and observed heterozygosity (Ho) values, HWE in terms of FIS coefficient and neutrality tests were estimated. Hence, in Korean Hanwoo the lowest number of alleles (8) was observed, whereas in Sheko the highest numbers of alleles (18) were detected. The observed heterozygosity (Ho) ranged between 0.750 in Fogera to 0.980 in Ethiopian Boran (Table 4). The expected heterozygosity (He) was ranged from 0.804 (Hanwoo) to 0.987 (Boran). Moreover, we recorded the following values of He from Red Chittagong (0.923), Fogera (0.975), Horro (0.965), and Sheko (0.979).
The Tajima’s D value obtained was negative in three breeds (Bangladesh, Horro, and Sheko), but Tajima’s D value in Boran, Fogera, and Hanwoo were 0.233, 0.350, and 0.203, respectively. Except for Korean Hanwoo breed which had Fu’s Fs recorded value of 4.594 all remaining breeds had a negative estimated value of Fs. Fu’s Fs p-values was highest in Hanwoo (0.959) breed and lowest in Sheko (0.052). Concerning the values of Hardy Weinberg equilibrium (HWE), all the breeds showed a significant deviation from the theoretical hypothesis (we estimated significant excess of homozygotes) (Table 5).
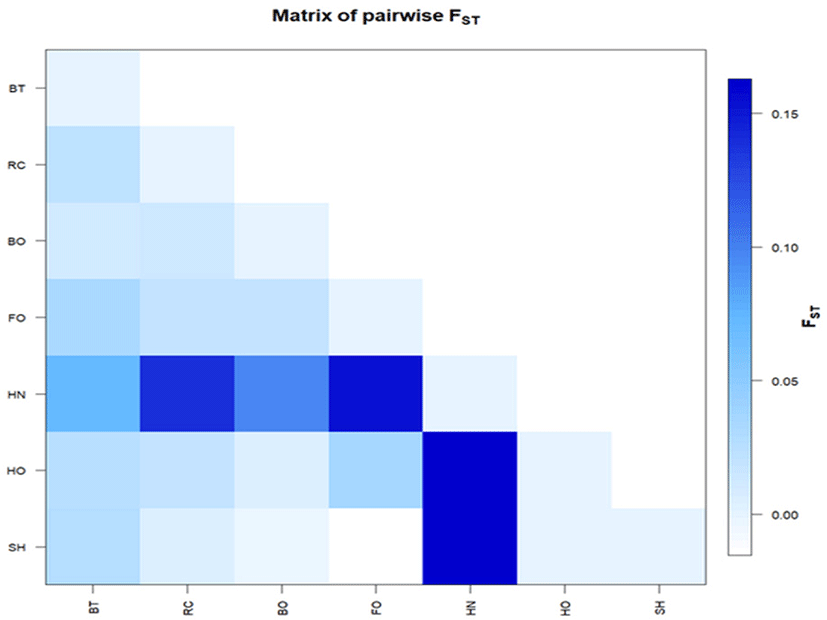
We estimated two main parameters (FST index and exact G test) for the inspection of population genetic structure and differentiation levels among cattle breeds. The FST parameters showed significant differences across all cattle breeds (FST = 0.056; p-value ≤ 0.0001); pairwise comparisons ranged from −0.003 (between Boran and Sheko) to 0.164 (Hanwoo with Horro and Sheko) (Table 5). The calculated FST values between population pairs were also graphically represented by using R-statistics (Fig. 3). The negative FST values recorded between Sheko and Boran as well as between Sheko and Fogera indicated that no genetic subdivision between these populations. The exact G test was compared between cattle breeds and the highest GST value was observed between Bangladesh Red Chittagong and Sheko (0.110). The lowest GST value (0.005) was found between Horro, and Boran (Table 5).
The evolutionary history was inferred by using maximum likelihood method based on Kimura-2 parameter model with bootstrap replications of 1000 and the evolutionary tree inferred from cattle for DRB3. 2 locus sequences are presented in Fig. 4. There was no clear separated cluster among the cattle breeds of this study, and they shared a common node from the constructed tree. It looks that DRB3 alleles might have evolved through multiple lineages.

DISCUSSION
The new alleles identified from the sequences of BoLA-DRB3 exon2 locus could function as additional marker specific to either Bos indicus or Bos taurus genotypes [29–33]. The biological response to pathogens and parasitic infection is strongly dependent on the level of genetic diversity at the MHC loci [34,35]. The allele BoLA-DRB3*009:02 was reported to be resistant to BLV [36] and detected in the Hanwoo breed (Table 2). In addition, the highest polymorphic sites and the total number of mutations of the BoLA-DRB3 gene were found in Sheko breed that could be a candidate for the future study of tsetse resistance.
Sequence analysis of the BoLA-DRB3 gene for base substitutions showed a higher rate of non-synonymous substitution (NSS) as compared to synonymous substitution (SS). The ratio of (NSS/SS) was evaluated and the values recorded were more than one for all the breeds. The result suggested that variation at the antigen-binding site (ABS) is under positive selection which could be for the recognition of a wide range of pathogenic agents [37,38]. The higher variability of the gene is highly linked to the regions of peptide binding sites (PBS) by which amino acid residues interacting directly with antigens. Therefore, molecules coded by different alleles have different antigen-binding profiles and again influence the susceptibility to pathogens [38]. Hence, the BoLA-DRB3 gene increased the fitness of the populations to live in pathogenic areas and have a positive impact in animal breeding. The widely reported studies indicated that Zebu cattle had high disease tolerance to tropical infectious diseases like tick born disease and intestinal parasites [39,40]. Therefore, the result of this study shared great insight and calls to the scientific community about the BoLA-DRB3 gene, especially for animal breeders and vaccine designers.
Investigating a population in terms of genetic diversity, researchers used nucleotide and haplotype diversities as common directories [41]. The values of heterozygosity were lower when compared with previous study of Tanzania Shorthorn Zebu, Boran, and Holstein Friesian breeds [30,42]. The heterozygosity in Japan cattle breeds were higher as reported by previous studies [43] (Ho ranged from 0.905 to 0.921 and He ranged from 0.887 to 0.914). Similarly, higher values of He were reported in Poland ranged from 0.920 to 0.927 [44]. The observed heterozygosity was not significantly higher than the expected heterozygosity (p < 0.05). Hence, HWE of all the breeds analyzed showed non-significant excess of heterozygotes animals that could reveal over-dominant selection. The result was similar with the recent report conducted to investigate South American Zebu breeds [7].
The results of pairwise FST values indicated low genetic differentiation of the Ethiopian breeds with Asian zebu which confirmed the investigation reported among Asian, African and American breeds [22]. The estimations of negative FST value between Sheko and Boran as well as between Sheko and Fogera, suggesting no population sub-division at this locus. Moreover, the genetic differentiation of all the breeds based on GST was small and supported by the previous studies [7,22,30]. The little genetic distance inferred from the breeds could be the result of the evolutionary or domestication history of cattle breeds [45].
Fu’s FS test [23] which was used to identify neutrality showed that all the breeds were neutral (p > 0.02). The result suggested that Ethiopian indigenous cattle breeds and Asian breeds are not significantly differentiated in terms of genetic diversity of the BoLA-DRB3 gene. The estimated negative values of Fu’s FS indicate the evidence for an excess number of alleles and could be expected from a recent population expansion or genetic hitchhiking. Whereas, positive values of FS deficiency of alleles, as would be expected from a recent population bottleneck. Except for the positive values of FS for Hanwoo we recorded the negative FS values from all the breeds. Ethiopian Sheko breed which is found in the Southern part of the Bench Maji zone was reported to be trypanotolerant as compared to zebu breeds. Due to the higher degree of trypanotolerance of the breed, Ethiopian Sheko cattle are kept in tsetse infested regions [46]. Breeds kept in the pathogenic environments are forced to develop adaptive immunity by the principles of natural selection and diversifications of immune-related genes could happen. Thus, we may have found the higher polymorphic sites and a greater number of total mutations rate in Sheko breed.
We observed a close genetic relationship between the Ethiopian cattle breeds and Asian breeds from the inferred phylogenetic tree and median joining-network tree. The close genetic relationship between Ethiopian cattle and Bangladesh Red Chittagong is consistent with the evidence of all Ethiopian cattle populations shared a higher proportion of Asian zebu ancestry [47–49].
This study confirmed that the MHC II DRB3.2 locus is extremely crucial and may provide baseline information for in-depth understanding and exploitation of MHC gene variation in Ethiopian, cattle populations exposed to an extensive range of pathogenic agents. The alleles we identified from the present populations require further validation to the association of disease susceptibility/resistance. Moreover, the sequence polymorphism of BoLA-DRB3.2 identified from our study should be further confirmed by sequencing more samples.