













INTRODUCTION
Lactobacillus acidophilus is a gram-positive, homofermentative, and microaerophilic bacteria. It ferments sugars into lactic acid and grows readily in acidic pH (below 5.0) and is common in the gastrointestinal (GI) tract of humans and other animals [1]. L. acidophilus is used in the production of fermented food and dairy products and is a symbiont in humans and animals. L. acidophilus strains are commercially used in many dairy products for the production of yogurt, health foods, and several medicines [2]. Some strains of L. acidophilus have probiotic characteristics. Several studies have shown that L. acidophilus exhibits various probiotic effects in humans and animals and helps in lowering cholesterol levels, preventing and treating diarrhea, modulating the immune system, and suppressing cancer [3,4].
Recently, there has been an increasing interest in developing methods to modulate the animal intestinal microbiota to improve health. The GI tract of healthy dogs contains Lactobacillus species, such as L. acidophilus [5]. Even after considering inter-individual variations, L. acidophilus is established in the gut of dogs soon after birth, similar to humans and other mammals. As it grows, it reaches compositional stability, with its principal activity being inhibition of undesirable microorganism proliferation [6]. Additionally, commensal gut bacteria positively interact with the host immune system, producing a wide range of metabolites crucial for host physiology. The gut bacteria depend on their host for nutrients and to maintain a stable ecosystem [7]. Therefore, the phylogenetic differentiation of L. acidophilus strains reflects their co-evolution with their vertebrate hosts. Previous studies have characterized the phylogenetic and genetic features of L. acidophilus strains isolated from diverse hosts using their genomic data [8, 9]. To date, dozens of strains isolated from several hosts have been studied and are published in the National Center for Biotechnology Information Center (NCBI). However, the phylogenetic and genetic features of L. acidophilus strains isolated from canine intestines remain unknown.
Recently, with the development of long-read and high-throughput DNA sequencing technologies, whole-genome studies have become increasingly feasible and affordable, making the genomic data of diverse organisms publicly available. Based on this data, comparative genomic analysis of strains within the same species has provided insights into modified, acquired, or lost genetic features closely related to the evolution and adaptation to specific environments [10]. In this study, we sequenced canine-derived L. acidophilus strain (C5) and performed comparative genomic analysis (pan-genome and the ratio of non-synonymous to synonymous substitutions [dN/dS] analysis) to profile its genetic characteristics.
MATERIALS AND METHODS
L. acidophilus C5 was isolated from dog feces (Shih Tzu, male) in Korea by Woogene B&G and was kindly provided for research purposes. L. acidophilus C5 was cultivated on modified de Man, Rogosa and Sharpe (MRS) (mMRS, with 0.05% cysteine-HCl) in an anaerobic atmosphere (5% hydrogen, 5% carbon dioxide, and 90% nitrogen) for 48 h at 37°C.
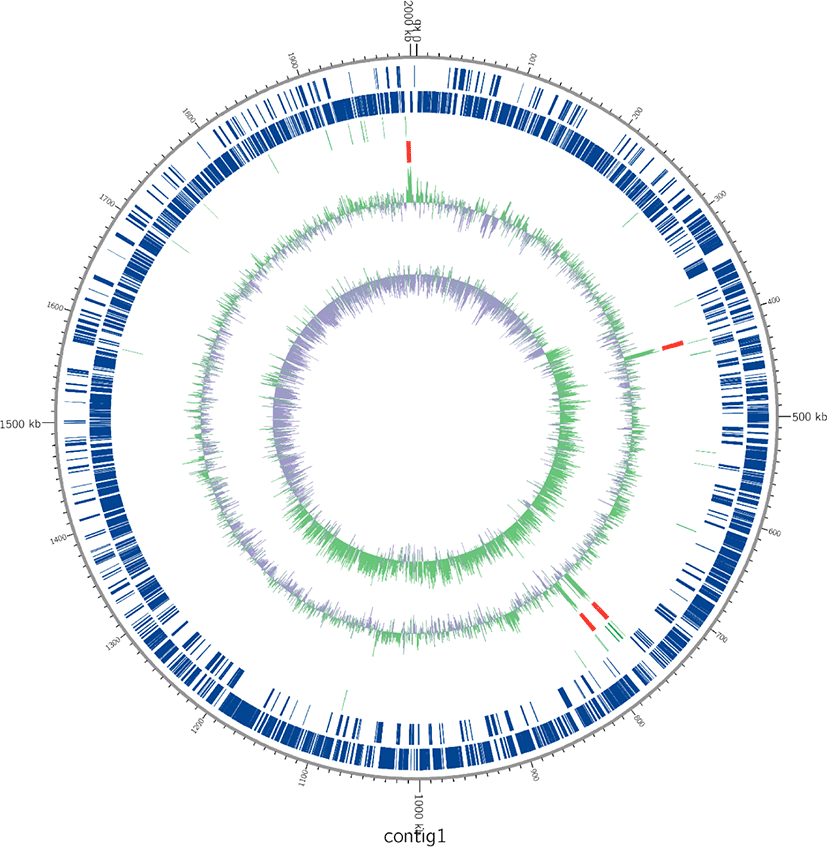
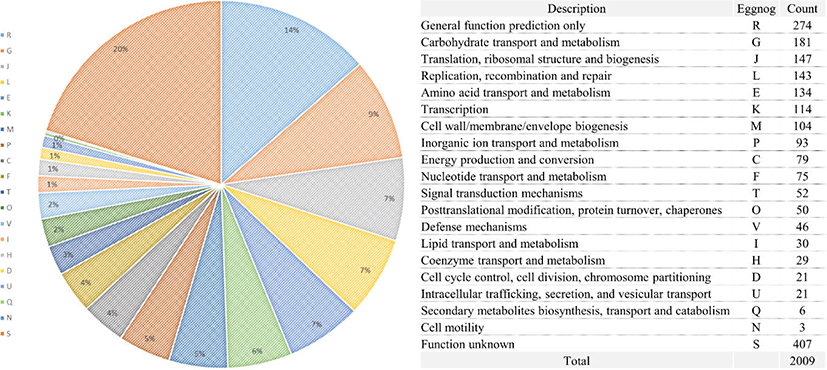
DNA was extracted using DNeasy UltraClean Microbial Kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions. Whole-genome shotgun sequencing of the L. acidophilus C5 strain was performed using the PacBio SMRT and Illumina HiSeq sequencing technology. For genome assembly, we applied the recently described Unicycler (v0.4.6) [11] and the final assembly was 2.005 Mb in one contig (Fig. 1). The PreAssembly step mapped single-pass reads to seed reads, the longest portion of the read length distribution. Subsequently, a consensus sequence of the mapped reads was generated, resulting in long and highly accurate fragments of the target genome. The next step was to correct and filter the reads. Reads that were fully contained in other reads did not provide extra information for constructing the genome, so they were filtered. Reads with an unsuitable extent of overlap were also filtered. Next, we constructed contigs of the L. acidophilus C5 strain. After de-novo assembly, we mapped the Illumina HiSeq reads to the first assembled genome sequence. We observed slight difference in the mapping result and the assembly result. We used this information to generate a consensus sequence of higher quality through a self-mapping step. Previously published genome sequences of six strains of L. acidophilus isolated from diverse hosts (yogurt, DuPont nutrition, and humans) were acquired from NCBI and compared with the host-derived strain. All genome sequences of L. acidophilus strains were annotated using Prokka (v1.12b) [12] and EggNOG-mapper (v5.0) [13]. The protein-coding sequences were categorized based on the Clusters of Orthologous Groups (COG) database from the Prokka results (Fig. 2). For comparative genomic analysis in this study, we selected strains with complete genome information in NCBI database. To evaluate the genetic relationship between L. acidophilus C5 and the other strains, the ANI was calculated using the JSpecies web server [14]. All information on L. acidophilus C5 and the other six strains used in this study is presented in Table 1 and Supplementary Table S1.
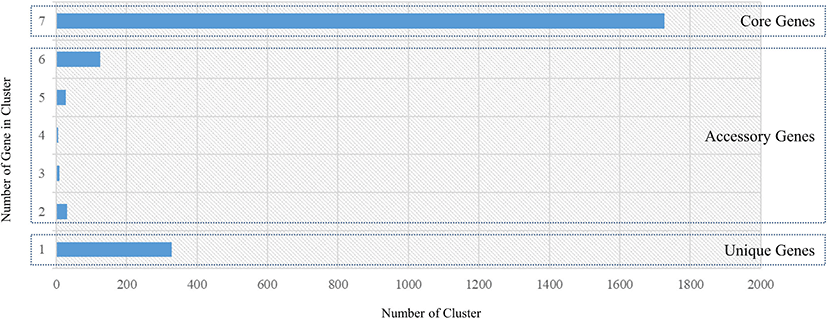
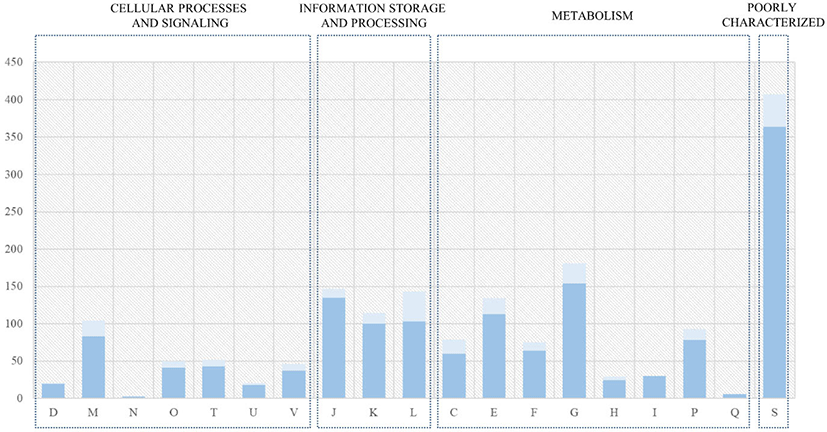
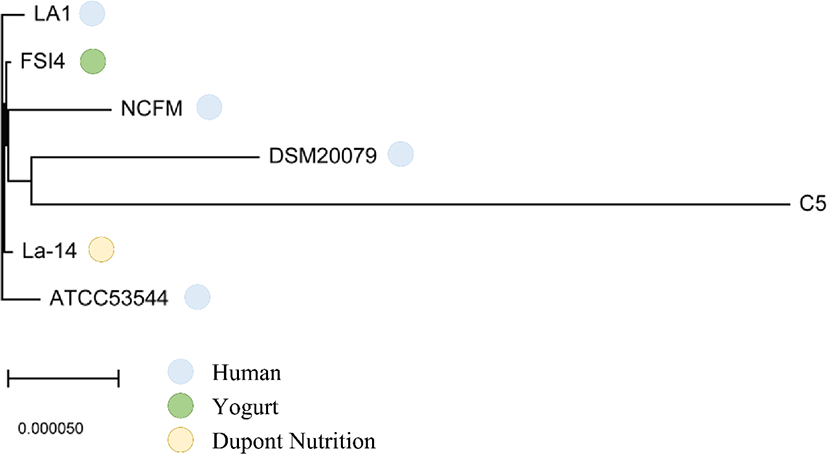
The genome sequences of seven L. acidophilus strains, including the C5 strain, were first annotated using Prokka (v1.12b) [12] to obtain GFF files, which were used to perform pan-genome analysis. The core- and pan-genomes were calculated using Roary, a rapid standalone pan-genomic pipeline [15] and a pair of genes was defined as belonging to the same gene family when the identity value of their amino acid sequences was > 95% (Fig. 3). The COG annotation (from EggNOG-mapper result) of core genes and the C5 strain is shown in Fig. 4. A phylogenetic tree was constructed based on the core genes (Fig. 5).
For comparative genome analysis using the dN/dS method, we used OrthoFinder (v1.1.10) [16] and PRANK [17] to determine ortholog genes for the seven genomes and multiple sequence alignment of each orthologous gene, respectively. These sequences were converted into the corresponding cDNA sequences using PAL2NAL [18] and poorly aligned transcripts were eliminated using Gblocks [19]. After all the filtering steps, a total of 1,843 orthologous groups remained. Phylogenetic analysis by maximum (PAML4) analysis using the maximum likelihood method [20] was used to estimate the dS and dN. Phylogenetically featured genes were investigated by the branch-site models.
RESULTS
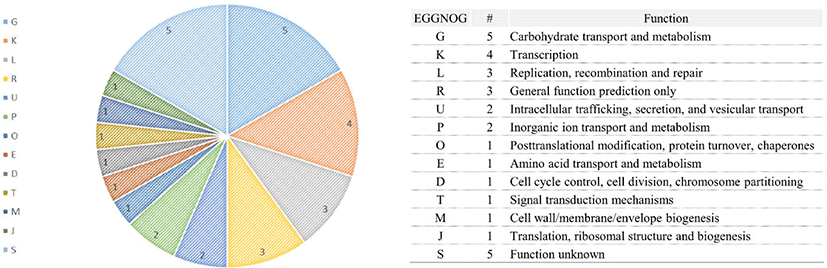
The total genome size of the L. acidophilus C5 strain in this study was 2.005 Mb, and the guanine + cytosine (G + C) content was 34.5% (after fitting). Additionally, genome annotation using Prokka (v1.12b) [12] and EggNOG-mapper [13] showed that the sequenced genome consisted of 2,009 coding genes and 73 non-coding genes (61 tRNA and 12 rRNA genes) (Fig. 1 and Table 1). In the functional analysis using the COG database in EggNog-mapper, the largest protein-coding categories (except, “General function prediction only” and “Function unknown”) in L. acidophilus C5 strain were “Carbohydrate transport and metabolism (G)” (9.01%), “Translation, ribosomal structure and biogenesis (J)” (7.32%) and “Replication, recombination, and repair (L)” (7.12%) (Fig. 2).
The core- and pan-genomes of the seven L. acidophilus strains, including the C5 strain, were analyzed using a comparative genomics method. The pan-genome of the seven L. acidophilus strains contained 2,254 gene families, and the core genome contained 1,726 gene families, indicating that together, the seven genomes were sufficient to represent the core genome of L. acidophilus. Moreover, the seven L. acidophilus genomes contained 200 accessory gene families (six isolates: 126 gene families, five isolates: 27 gene families, four isolates: 5 gene families, three isolates: 10 gene families, and two isolates: 32 gene families), and 328 strain-specific genes (Fig. 3). To determine the functions of the 1,726 core genes, we extracted the sequences of the core genes to map to the COG database. The results showed that except for the “General function prediction only” and “Function unknown” categories, the largest proportion of core genes belonged to “Carbohydrate transport and metabolism (G, 154 genes)” followed by “Translation, ribosomal structure and biogenesis (J, 135 genes)”, and “Amino acid transport and metabolism (E, 113 genes).” (Fig. 4). Using a phylogenetic tree based on core genes, we found that the L. acidophilus C5 strain was clearly distinguished from the other six strains (Fig. 5). The strains closest to the L. acidophilus C5 strain were the human-derived strains (DSM20079 and NCFM). The C5 strain had the highest number of unique genes (245 genes) among the seven L. acidophilus strains. Among the unique genes of the C5 strain, the largest proportion belonged to “Replication, recombination, and repair (L) (28 genes)” and “Carbohydrate transport and metabolism (G) (24 genes)” apart from the “Function unknown” category (Fig. 4).
We performed dN/dS analysis (branch-site model) to identify the evolutionarily selective genes in the L. acidophilus C5 strain. Considering the phylogenetic relationships among the seven L. acidophilus strains, we searched for the genes that could explain the specific characteristics of the L. acidophilus C5 strain. We identified 1,843 orthologous genes from the seven strains and measured the rate of evolution using the dN/dS analysis (). We identified 30 phylogenetically featured genes and the variations in their amino acid sequences (Supplementary Table S2). To determine the functions of the 30 evolutionarily selective genes, we mapped their sequences to the COG database (Fig. 6). We observed that apart from the “Function unknown” category, the largest proportion of core genes belonged to the “Carbohydrate transport and metabolism (G)” and “Transcription (K)” categories. The carbohydrate transport and metabolism (G) category included five genes- C5_1_01133 (glcU_1: glucose uptake protein GlcU), C5_1_00898 (fba: fructose-bisphosphate aldolase), C5_1_01372 (hypothetical protein), C5_1_01889 (ptsI: phosphoenolpyruvate-protein phosphotransferase), and C5_1_00253 (malL_2: Oligo-1,6-glucosidase).
DISCUSSION
This study is the first attempt to decipher the genetic features and evolutionary adaptations of L. acidophilus in the canine gut intestine. We performed whole-genome sequencing to construct the genome of the L. acidophilus C5 strain and compared the genomic information of L. acidophilus derived from a dog with six other strains isolated from diverse hosts. We determined the genetic basis for the characteristics of the L. acidophilus C5 strain that are likely related to the host. After the sequencing and assembly process, we were able to construct one large contig corresponding to the genome of the C5 strain, comparable in size (2.005 Mb) with the other six complete genomes of L. acidophilus (NCFM, LA14, FSI4, ATCC53544, LA1,and DSM20079) from NCBI (Table 1) [21–26]. In this study, complete genome sequences of other strains in the NCBI database were used for comparative genome analysis of C5, which were isolated from different hosts, and none were isolated from dogs as in previous studies [27,28]. In the annotation process, we found 2,082 genes (2,009 coding and 73 non-coding genes), which was slightly more than the number of genes found in the other six strains. Similarly, the genome size of L. acidophilus C5 was slightly larger than that of the other strains. In the functional analysis using the COG database, it was found that genes related to carbohydrate transport and metabolism (181 genes) comprised the largest part of the L. acidophilus C5 strain genome, apart from “General function prediction only” and “Function unknown” categories. In other strains too, the carbohydrate metabolism-related genes comprised a large portion of the genome (Fig. 4), but C5 was the only strain in which they were most abundant among the categories with distinct functions (except for “General function prediction only” and “Function unknown” categories).
In pan-genome analysis, a total of 1,726 core genes were detected in the seven L. acidophilus genomes isolated from four hosts, which mainly encoded essential proteins for metabolism (30.35%) (Fig. 4). Consistent with the findings of studies on other Lactobacillus strains, our findings suggested that core genes are indispensable, constitute the basic framework of the L. acidophilus, and play important roles in carbohydrate transport and metabolism (154 genes) and translation, ribosomal structure, and biogenesis (134 genes). Competitive retention of L. acidophilus in the intestinal tract is important for glycogen biosynthesis in carbohydrate metabolism, which demonstrates that the ability to synthesize intracellular glycogen contributes to gut fitness and indicates retention of probiotic microorganisms [29,30]. After pan-genome analysis, we constructed a phylogenetic tree based on the 1,726 core genes to evaluate the genetic relationships between the canine C5 strain and the six other strains of L. acidophilus. In this phylogenetic tree, we identified that C5 was distant from other strains but was close to two strains (DSM20079 and NCFM) from humans. This could be attributed to the fact that dogs are representative companion animals to humans and have shared their environments for a long time. Therefore, the genomic differences in L. acidophilus strains and similarities between the C5 and the two strains from humans might be associated with their colonized environments. Significantly, the strains closest to C5 were human-derived even if all human-derived strains were not close (such as LA1 and ATCC53544). Similar results were obtained in a comparative genome study of other Lactobacillus strains [27]. To better understand the characteristics of L. acidophilus C5, we investigated the unique genes of all L. acidophilus strains in this study. From the 2,254 gene clusters in pan-genome analysis, we found 328 unique genes, 245 of which were specific to the C5 strain. In the functional analysis based on the COG database, these C5-specific genes were mainly associated with “Replication, recombination, and repair (28 genes)” and “Carbohydrate transport and metabolism (24 genes).” Interestingly, core gene analysis revealed many common carbohydrate metabolism-related genes in the L. acidophilus strains, and it was confirmed that many carbohydrate metabolism-related genes in the C5 strain were not found in other strains. Moreover, we found that there were more carbohydrate metabolism-related genes in the C5 strain than in the other strains. Based on the core gene analysis and unique gene results, we inferred that C5 has distinct genomic features from other strains of L. acidophilus.
The similarity of several genomes within or between species is the basis of comparative genomics. If two species or strains have a recent common ancestor, the differences between the two genomes evolved after the common ancestral genome. The more closely related the two strains, the higher the similarities between their genomes [31]. When we performed comparative genomic analysis of the C5 strain isolated from dogs with other strains of L. acidophilus, we supposed that the genomic differences between the L. acidophilus strains were significant in the evolutionary process and could explain the adaption process of the C5 strain using the evolutionary statistical method (dN/dS) [32]. The identification of genetic loci undergoing adaptation is a central aim of evolutionary biology, and several statistical tests have been developed to quantify selection pressures acting on protein-coding regions. Among these methods, the dN/dS ratio is one of the most widely used. It is simple and robust and can quantify selection pressures by comparing the rate of substitutions at silent sites (dS, which are presumed neutral) to the rate of substitutions at non-silent sites (dN, which possibly experience selection). The dN/dS ratio is used for distantly diverged sequences, so the differences among them represent substitutions that have been fixed along independent lineages [33,34]. We assumed that the selection signal by dN/dS indicated adaptation of each strain to its environment and used this measurement to understand the adaptation of the C5 strain to the canine environment [28, 35]. After identifying 1,843 orthologous genes in the seven genomes, we performed dN/dS estimation for each orthologous gene using PAML4 [20]. We determined 30 evolutionarily selective genes for the C5 strain. In these 30 genes, there were 300 C5-specific amino acid changes, out of which 141 were statistically significant. In the crcB gene, there were 30 C5-specific amino acid changes, the highest among the 30 selective genes, and 14 of all amino acid changes were statistically significant. This gene is important for preventing fluoride toxicity by reducing its concentration in the cell [36]. In the functional analysis based on the COG database, these evolutionarily selective genes were mainly associated with carbohydrate transport and metabolism (five genes) and transcription (four genes). Interestingly, among the genes containing significant evolutionary selection signals, most genes were carbohydrate metabolism-related genes. We hypothesized that the genes related to carbohydrate metabolism in strains isolated from dogs were closely related to the domestication of dogs. The domestication of dogs was an important milestone of human civilization [37]. In a previous study, whole-genome re-sequencing of dogs and wolves was performed to identify genomic regions potentially representing selection targets during dog domestication [38]. This study identified candidate mutations in key genes and provided functional support for increased starch digestion in dogs relative to wolves. This result indicates that adaptations allowed modern dog ancestors to survive on a diet rich in starch, a crucial step in the domestication of dogs. We wondered whether the evolution of L. acidophilus C5 from dogs has occurred. Reportedly, the dog and human gut microbiomes are similar in terms of gene content and response to diet [39]. As dogs and humans have shared a similar environment for a long time after domestication, if the dogs experience similar metagenome changes due to changes in diet, it was deduced that its microbiome adapts to a carbohydrate-rich diet. We inferred that more carbohydrate-related genes in the genome and genes with selection signals in L. acidophilus C5 were the result of evolution and domestication.
In summary our results indicated that the L. acidophilus C5 strain from a canine host had many genes related to carbohydrate metabolism, presumably due to domestication by humans. We reported the characteristics of the C5 strain from a canine using whole-genome sequencing data compared with other original isolates, providing a strong indication of the factors affecting its evolutionary history (evolution due to domestication of dogs by humans). We hope that our study contributes to the feasibility of using these strains as probiotics for dogs in the future.