













INTRODUCTION
There is a large and complex microbial community in the gut, of which over 1000 bacterial species have been identified, some of which have not yet been classified [1]. The microbiota can metabolize nutrients that the host cannot digest, and some species increase digestibility through this process and provide the host with energy sources, such as short-chain fatty acids [2]. Another important function of the intestinal microbiota is the development of the intestinal mucosa and its immunomodulatory function. Given that the intestinal mucosa is a defense line that blocks external antigens, maintaining its integrity is considered immensely important for the immune system [3]. In addition, various immune cells exist on the intestinal surface, and an immune response is constantly generated due to interactions with foreign substances. The intestinal microbiota, particularly some bacterial species, plays an important role in this process [4]. As the significance of the intestinal microbiota is emphasized, the exploration of the intestinal microbial repertoire and interactions with the host is increasing rapidly [5,6].
Numerous studies on the relationship between intestinal microbes and host conditions and diseases have been conducted using metagenomics [7,8]. However, culture-independent analysis is biased based on DNA extraction protocols or differences in primers used for amplification [9] and cannot detect minor-level populations [10]. In addition, metagenomics analysis cannot provide information regarding mechanisms and the role of bacteria due to a lack of securing culturable microbiota. In particular, given that bacteria are not recovered, there is a limit to the use of in vitro/in vivo experiments or probiotic bacteriotherapy [11]. As the limitations of metagenomics have been revealed and the importance of culturomics has emerged, a wide range of culture-dependent analyses of intestinal microbiota and studies on optimal culture methods for gut microbiota are being conducted [10,12–15]. However, most of the studies focused exclusively on the analysis of human gut microbiota, and studies on culturomics of fecal microbiota of livestock animals are extremely rare [16,17].
Pigs are monogastric animals and have physiological and immunological characteristics similar to humans. In addition, pigs have gastrointestinal (GI) tract similar in size to humans and share the same constitutive features of intestinal microbiota with similar omnivores, such as humans [18,19]. From the perspective of the livestock industry, the importance of exploring the characteristics of intestinal microbiota is growing rapidly as interest in animal health and welfare is increasing in contrast to studies simply pursuing production efficiency [20,21]. However, most studies on the intestinal microbiota of pigs are focused on culture-independent analysis, and culturomics-based research has rarely been conducted [22,23].
Therefore, this study was conducted to distinguish the structure of sow and piglet microbiota using both culture-independent and culture-dependent viewpoints, to understand the mechanism of the role of intestinal microbes and to lay a foundation for its potential for use as a microbial treatment.
MATERIALS AND METHODS
The culturomics process is shown in Fig. 1A. Fresh fecal samples of sows and weaned pigs were collected from 3 pig farms located in 3 different provinces in Korea, and immediately placed in sterile plastic tubes under anaerobic conditions with the GasPak EZ anaerobe pouch system (Becton Dickinson, Franklin Lakes, NJ, USA) until transfer to an anaerobic chamber (COY Laboratories, Grass Lake, MI, USA). Fecal samples for metagenomic analysis were collected directly from each pig with sterile cotton swabs, transported to the laboratory on ice and placed at –80°C in a deep freezer until further analysis. Inside the COY anaerobic chamber, fecal samples were homogenized and serially diluted with 0.85% (w/v) NaCl solution, and a 100-μL aliquot from the dilution was spread on agar plates of two nonselective media including phenylethyl alcohol agar (PEA; MB Cell, Seoul, Korea) and fastidious anaerobe agar (FAS; Acumedia, Neogen, Lansing, MI, USA) and two selective media such as de Man Rogosa Sharpe agar (MRS; BD) for lactobacilli and Bifidobacterium selective agar (BS; MB Cell) for bifidobacteria spp. After plating, plates were incubated in an anaerobic chamber at 37°C for 48 h. Single colonies were selected and subcultured at least thrice under their respective culture conditions to obtain pure isolates. Isolated bacteria were stored at −80°C in cryovials containing 250 μL of 50% glycerol and 750 μL of bacterial culture. The procedure of experiment was reviewed and approved by the Institutional Animal Care and Use Committee of Chungnam National University, Daejeon, Korea (approval #CNU-00910). All animal handing and sampling procedures in this study followed the guidelines and regulations for the animal use.

The bacterial cultures were centrifuged to acquire cell pellets. Total genomic DNA was extracted using the PureHelix™ Genomic DNA Prep Kit (Nanohelix, Daejeon, Korea) following the manufacturer’s instructions with minor modifications. For molecular identification using 16S rRNA sequencing, universal primer set 27F-1492R was used to amplify the 16S rRNA gene fragments, and the amplified product was sequenced using an ABI 3730xl DNA analyzer (Applied Biosystems, Foster City, CA, USA). The 16S rRNA gene sequence was analyzed with GenBank BLASTn using the Complete Prokaryote Genome Database for taxonomic annotations of isolated bacteria. Sequences with a percent similarity less than 98.7% were defined as new bacterial species, and those less than 95% were defined as novel bacterial genera [24].
For comparing with culturomic approach, fecal sample DNA was extracted for metagenomic analysis using a DNeasy PowerSoil Pro kit (Qiagen, Hilden, Germany) according to the provided manufacturer’s protocol. After extraction, the amount and quality of DNA were assessed by measuring absorbance using a spectrophotometer (SpectraMax ABS Plus, Molecular Devices, San Jose, CA, USA). Amplification of the V4 region of the 16S rRNA gene was performed (primer set: forward, 5′-TCG TCG GCA GCG TCA GAT GTG TAT AAG AGA CAG GTG CCA GCM GCC GCG GTA A-3’; reverse, 5′-GTC TCG TGG GCT CGG AGA TGT GTA TAA GAG ACA GGG ACT ACH VGG GTW TCT AAT-3′), and amplicon sequencing was conducted at Macrogen (Seoul, Korea) using the Illumina MiSeq platform (Illumina, San Diego, CA, USA). The obtained paired-end sequence was analyzed using Quantitative Insights Into Microbial Ecology 2 (QIIME2; version 2020.6.0). Quality control of sequence reads (e.g., filtering, trimming sequence and denoising) was performed with the DADA2 pipeline. Taxonomic assignment into operational taxonomic units (OTUs) with 97% similarity was performed using the Greengenes reference database (13_8 version).
RESULTS
To increase the diversity of anaerobic isolates, both selective and nonselective media were employed. From the sow and weaned pig fecal samples, a total of 278 and 149 colonies were obtained, respectively (Fig. 1B). Isolated bacteria from sow fecal samples were classified into 19 species from four phyla, including Firmicutes (77.26%, 14 species), Actinobacteria (13%, 2 species), Proteobacteria (6.86%, 1 species), and Bacteroidetes (2.89%, 2 species). These species were divided into nine different genera, and the five most isolated genera included Lactobacillus (72.66%), Bifidobacterium (12.95%), Escherichia (6.83%), Bacteroides (2.88%), and Bacillus (2.16%). Among the isolated species, L. ruminis (32.73%) was the dominant species followed by L. amylovorus (25.18%), L. mucosae (8.99%), B. thermophilum (6.83%), and Escherichia coli (6.83%). A total of 12 bacterial species were obtained from weaned pig fecal samples, and isolates were classified into four phyla: Firmicutes (56.08%, 8 species), Proteobacteria (33.78%, 1 species), Actinobacteria (9.46%, 2 species), and Bacteroidetes (0.68%, 1 species). Isolated bacterial species were categorized into seven genera, and Lactobacillus (36.91%), Escherichia (33.56%), Enterococcus (17.45%), Bifidobacterium (8.05%), and Olsenella (2.01%) represent the top 5 species. E. coli (32.89%) predominated among the colonies obtained from piglet feces followed by L. ruminis (16.11%), E. cecorum (16.11%), L. reuteri (15.44%), and B. thermophilum (7.38%).
The bacterial genera isolated from both sows and piglet fecal samples included Acidaminococcus, Bacteroides, Bifidobacterium, Enterococcus, Escherichia, and Lactobacillus for a total of 6 genera, and Lactobacillus was the genus most often isolated in both samples. Of these genera, Acidaminococcus and Bacteroides accounted for less than 3% of both samples. Lactobacillus accounted for greater than 70% of the bacteria isolated from sow feces followed by Bifidobacterium at 12.95%. However, in piglet feces, Lactobacillus accounted for approximately 37%, and Escherichia and Enterococcus accounted for approximately 34% and 17%, respectively. In addition, Aerococcus, Bacillus, and Erysipelotrichaceae gen. were isolated only from sow samples, and the Olsenella genus was isolated only from fecal samples of weaned pigs. In addition, according to 16S rRNA sequence analysis, 1 genus and 15 new bacterial species were discovered in sow feces, and 3 new bacterial species were obtained in piglet feces.
The isolated number of the corresponding bacterial species in the culture medium of each sample was expressed as a heat map (Fig. 1C). Comparing the number of bacterial species isolated by the medium, 16 and 6 species in PEA medium were identified in sow stool and weaned pig stool, respectively. In the FAS medium, 10 species of sow fecal isolates were identified, and 8 species of bacteria were isolated from the feces of weaning piglets. In sow fecal samples, a total of 6 and 3 bacterial species were isolated in MRS and BS media, respectively. In piglet fecal samples, 7 bacterial species were isolated from MRS medium, and 5 bacterial species were isolated from BS medium. Thus, the number of species isolated from each medium was different, but the percentage of total isolated colonies was 29.86% (n = 83), 28.42% (n = 79), 19.42% (n = 54), and 22.3% (n = 62) in sow feces in each medium, and 29.53% (n = 44), 27.52% (n = 41), 23.49% (n = 35), and 19.46% (n = 29) in piglet feces, respectively.
To compare the results of culture-dependent and culture-independent analyses, metagenomics of the same sample used for culturomics was performed. Both fecal samples generated a total of 609,576 reads. After quality control and removal of chimeras in OIIME2, the sequences were clustered into 581 OTUs based on 97% identity in fecal samples. The top 10 results based on relative abundance of fecal microbiota of sow and weaned pig at phylum and genus level are shown in Fig. 2. The bacterial composition of the sow fecal sample at the phylum level was composed of Firmicutes (48.61%) followed by Bacteroidetes (23.99%), Spirochaetes (20.42%), Proteobacteria (3.07%), Actinobacteria (0.64%), and Fibrobacteres (0.12%). Compared to the sow sample, the fecal microbiota of weaned pigs was classified into eight phyla: Spirochaetes (40.26%), Firmicutes (37.65%), Bacteroidetes (16.73%), Actinobacteria (0.46%), Proteobacteria (0.34%), Fibrobacteres (0.06%), Candidatus Melainabacteria (0.01%), and Chlamydiae (0.01%). The abundances of unclassified phyla in the sow and piglet fecal microbiota were 3.16% and 4.48%, respectively. A total of 98 genera were classified from sow samples with Treponema (20.4%), Prevotella (8.88%), Christensenella (8.42%), Muribaculum (7.12%), and Turicibacter (5.33%) in the top 5. By comparison, 104 genera were identified in weaned pig feces, and the 5 most abundant genera included Treponema (40.25%), Clostridium (18.45%), Prevotella (7.7%), Lactobacillus (3.60%), and Barnesiella (2.92%).

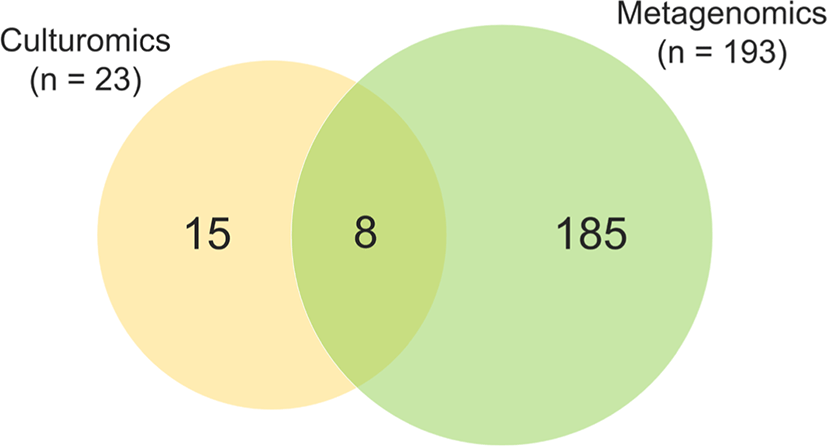
In this study, a total of 206 bacterial species and 2 archaeal species were identified in two fecal samples through culture-dependent and culture-independent analyses. In detail, a total of 23 bacterial species were isolated through the culture-dependent approach, and 191 bacterial species and 2 archaeal species were detected through culture-independent analysis. Interestingly, of the 23 bacterial species cultured in this experiment, 65% (n = 15) were not detected by metagenomics analysis (Fig. 3). Bacteria detected exclusively by culturomic approach including Bacillus genus (B. glycinifermentans, B. paralicheniformis, B. licheniformis, and B. thermoamylovorans) and other advanced features (Aerococcus urinaeequi, Erysipelotrichaceae gen., Bacteroides fragilis, Bacteroides vulgatus, B. pseudolongum, B. thermophilum, E. avium, E. faecalis, E. faecium, E. coli, and L. amylovorus).
DISCUSSION
Although numerous studies on the composition and diversity of porcine gut microbiota and their association with health state have been conducted over the past decades, studies using a culturomics approach to assess the pig gut microbiota are extremely rare. In addition, no studies have been conducted to compare the intestinal microbiota of sows and weaning pigs by culturomic analysis. In this study, to gain insight into the composition and functional characteristics of the intestinal microbiota of sows and weaned pigs, the microbiota of each fecal sample was evaluated by combining culture-dependent and culture-independent approaches. Of the 10 genera that accounted for a high proportion in the metagenomics results, none of the genera were recovered except for Lactobacillus. In addition, most of the genera isolated by culture-based methods appear to be represented excessively when compared to those identified by metagenomics approaches. This result suggests that the proper integration of metagenomics and culturomics is necessary for a complete understanding of intestinal microbiota.
Among the isolates recovered through culturomic analysis, Lactobacillus spp., which account for the highest proportion in both samples, are regarded as the core genera of healthy pigs according to previous studies and are the most commonly used probiotic bacteria [25]. In particular, a positive correlation was observed between Lactobacillus spp. and feed efficiency, and a significant correlation with growth performance was also reported [26]. Bifidobacterium is one of the most commonly used probiotic bacteria along with Lactobacillus [27]. Both culturomic analysis and metagenomic analysis showed that Bifidobacterium occupies a higher content in sows’ gut microbiota than in piglets. According to previous studies, Bifidobacterium is one of the major constituents of the intestine of sows [28], and Bifidobacterium has been associated with an increase in nutrient digestion in the later stages of pregnancy in sows [29].
One of the bacterial species identified exclusively using culturomics, Aerococcus urinaeequi, is a facultative anaerobic strain that was reclassified from Pediococcus urinaeequi to the Aerococcus genus in 2005 and is characterized by the production of lactic acid [30]. In addition, this strain inhibits the growth of Gram-negative pathogenic bacteria by producing bacteriocin in aquaculture [31]. However, further mechanistic studies are needed to determine how Aerococcus urinaeequi affects the host in the GI tract. The Erysipelotrichaceae gen. isolated in this study is a novel genus with a percent similarity less than 95%. This genus has not yet been classified, and further bacterial characterization studies are required [32–34].
Previous studies on culturomics have isolated hundreds of bacterial species using a variety of media and supplements [35,36]. However, using dozens of culture media is expensive and time consuming. Therefore, although fewer bacteria were isolated than noted in previous studies, several commercially available selective and nonselective media were used. Selection of the optimal culture medium that costs less and requires less time while being able to isolate various bacterial species is considered essential. In addition, although the culture-dependent analysis of microbiota remains limited to fecal microbiota, culture-based microbial analyses based on animal species, health status, or growth stage are needed to understand the interaction between livestock health and microbiota and dissect the mechanism of action.
In conclusion, in this study, two perspectives of microbiota analysis were conducted with fecal samples from healthy sows and weaned pigs. Although the culturomic approach is not suitable for grasping the composition of the entire microbial community at a glance, this approach has been able to confirm that intestinal microbial populations exist that cannot be identified by metagenomics. In addition, by recovering living bacteria through this experiment, we laid the foundation to confirm the types of interactions and mechanisms of action of intestinal microbes with the host.