
INTRODUCTION
Lumpy skin disease (LSD) in cattle and buffalo is caused by the LSD virus belonging to the family Poxviridae and genus Capripoxvirus [1–3]. Mosquitoes, especially Aedes aegypti, can transmit the LSD virus for at least 6 days without significant loss of titer [3,4]. The incubation period for LSD is approximately 7 days. The main symptom is sporadic swelling of the skin, with the appearance of nodules having diameter 0.5–5 cm [3,5,6]. Other symptoms include high fever of > 40°C, rapid reduction in milk production, loss of appetite, nasal discharge, salivation, swollen lymph nodes, weight loss, miscarriage, and infertility [1–3,5,7–11].
LSD was first reported in 1929 in Zambia, from where it spread to numerous places, including South Africa, North Africa, the Middle East, Europe, and Asia [1,3,12–14]. According to a recent report by the World Animal Health Information System and the Ministry of Agriculture, Food and Rural Affairs (MAFRA), Republic of Korea, LSD first broke out in Republic of Korea in Seosan city on October 20, 2023.
To prevent the spread of LSD, the immediate slaughter of all cattle that have come in contact with infected cattle and elimination of the initial source of infection are recommended [3,12,15].
However, if the disease has already spread widely, vaccination is recommended in most countries because this is the only method of prevention [6,15]. According to a report by MAFRA, 3 types of LSD vaccines were used in Republic of Korea. As of 14:00 on November 5, 2023, the status of LSD vaccination in Korea was 90.9% (3,766,000/4,076,000 cattle). To complete the nationwide vaccination by November 10, 2023, cows in areas at risk of LSD are being vaccinated by city/county vaccination groups (2,065 people from 931 classes nationwide) and farm owners (self-vaccination).
Studies have shown that cows demonstrate fever (83.9%), decreased feed intake (85.9%), and reduced milk production (94.6%) when the LSD vaccine is administered [16]. The analysis of changes in rectal temperature according to the LSD vaccination showed that compared with that of the control animals, the rectal temperature increased, and high-dose vaccinations resulted in rise of temperatures to ≥ 40°C [11]. Other studies showed that the highest rectal temperature was recorded 8 days after LSD vaccination, and milk production decreased by up to 16% [17].
While vaccination is required to prevent LSD, studies comparing body temperature and activity in pregnant and non-pregnant cows have not been conducted to date. Therefore, this study aims to analyze the patterns of changes in ruminoreticular temperature and body activity measured using a bolus sensor after LSD vaccination in pregnant and non-pregnant Hanwoo cows.
MATERIALS AND METHODS
The cows used in this study were bred at the Gyeongsangbuk-do Livestock Technology Research Institute, fed according to the Korean Feeding Standard for Hanwoo, and housed in pens (rearing space = 300 m2/15 cows) equipped with stanchions. Before beginning the experiment, cows with no abnormalities in the ovaries and uterus were selected by ultrasound examination. Finally, 46 cows (18 pregnant cows, 28 non-pregnant cows) were chosen for the study.
All the experiments were approved by the Animal Ethics Committee of the Gyeongsangbuk-do Livestock Research Institute (approval number: protocol code GAEC/140, approval date: December 14, 2021). Table 1 shows the age, parity, and pregnancy day of the cows used in the experiment.
| Group | Number of cows | Age of months | Parity | Days of pregnancy |
|---|---|---|---|---|
| Non-pregnant cows | 28 | 51.2 ± 4.0 | 1.8 ± 0.1 | 173.6 ± 3.7 |
| Pregnant cows | 18 | 47.0 ± 3.5 | 1.6 ± 0.2 | |
| Total | 46 | 49.7 ± 2.9 | 1.7 ± 0.1 |
Six months before beginning the experiment, a bolus sensor (smaXtec, Tauranga, New Zealand) was orally administered and placed in the cow’s rumen or reticulum; the adaptation period was 6 months. Information regarding the sensor used in the experiment and the method of measuring temperature and activity in the rumen every 10 min have been described in detail previously [18].
LSD vaccine (Lumpyvax®, Republic of South Africa; each 1 mL [1 dose] of the vaccine contains 104 TCID50 of freeze-dried, live, attenuated virus) was administered after disinfecting the vaccination site using 70% alcohol. The powder was dissolved in the dilution solution and subcutaneously injected (1 mL/cow) into the neck of the cow using a disposable syringe.
RESULTS
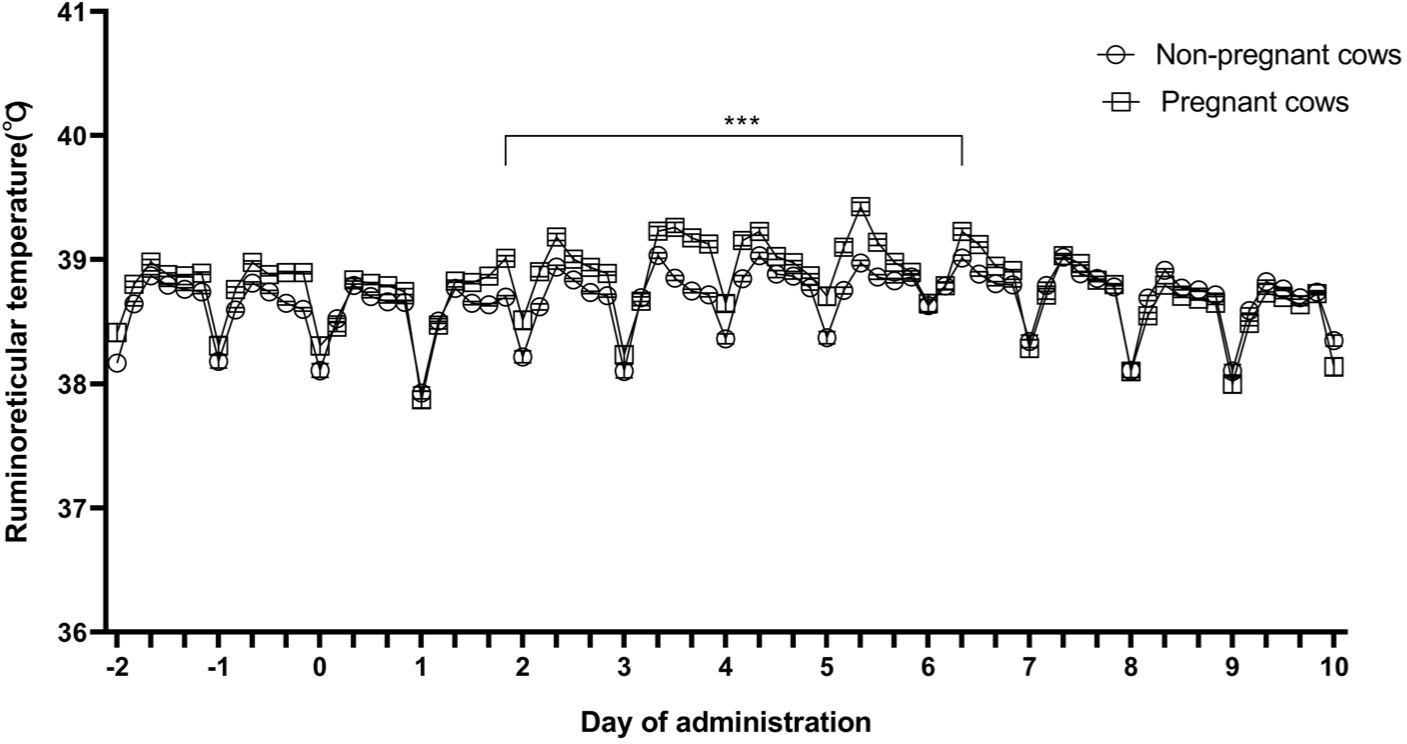
The ruminoreticular temperatures of pregnant and non-pregnant cows were measured at 10-minute intervals before and after LSD vaccination using the sensor. The average temperature for 4 hours is shown in Fig. 1. Two days before LSD vaccination, the average ruminoreticular temperatures of the pregnant and non-pregnant cows were 38.89 ± 0.01°C and 38.74 ± 0.01°C, respectively (Fig. 1). Two days after LSD vaccination, the ruminoreticular temperatures in both the groups gradually increased; this continued until 6 days after vaccination (p < 0.001). The rise in ruminoreticular temperature was greater in pregnant cows than it was in non-pregnant cows 3–5 days after vaccination (p < 0.001).
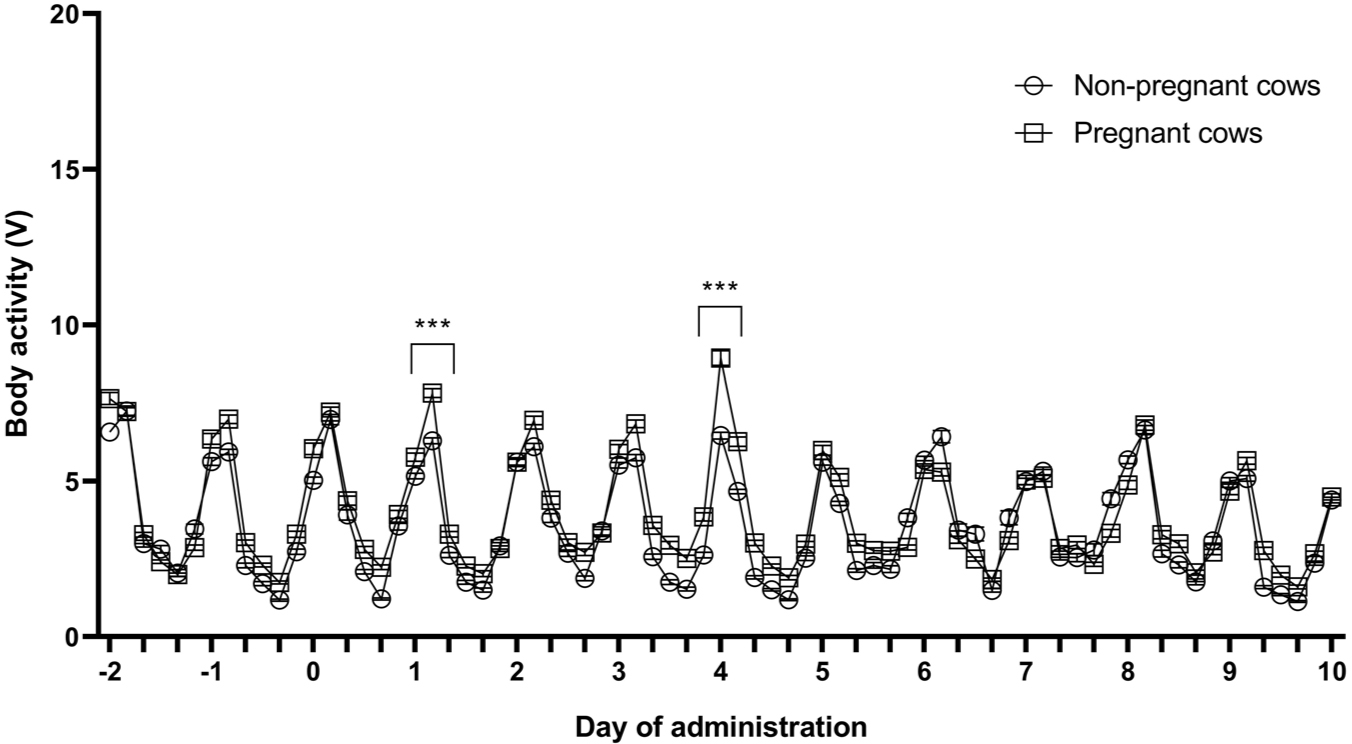
The body activity of pregnant and non-pregnant cows were measured at 10-minute intervals before and after LSD vaccination using the sensor. The average body activity for 4 hours is shown in Fig. 2. Two days before LSD vaccination, the mean body activity of the pregnant and non-pregnant cows were 2.41 ± 0.06 V and 2.81 ± 0.12 V, respectively (Fig. 2). No significant difference in the body activity of pregnant and non-pregnant cows was observed before and after LSD vaccination. However, the body activity of pregnant cows temporarily increased 1 and 4 days after vaccination compared with that in non-pregnant cows (p < 0.001).
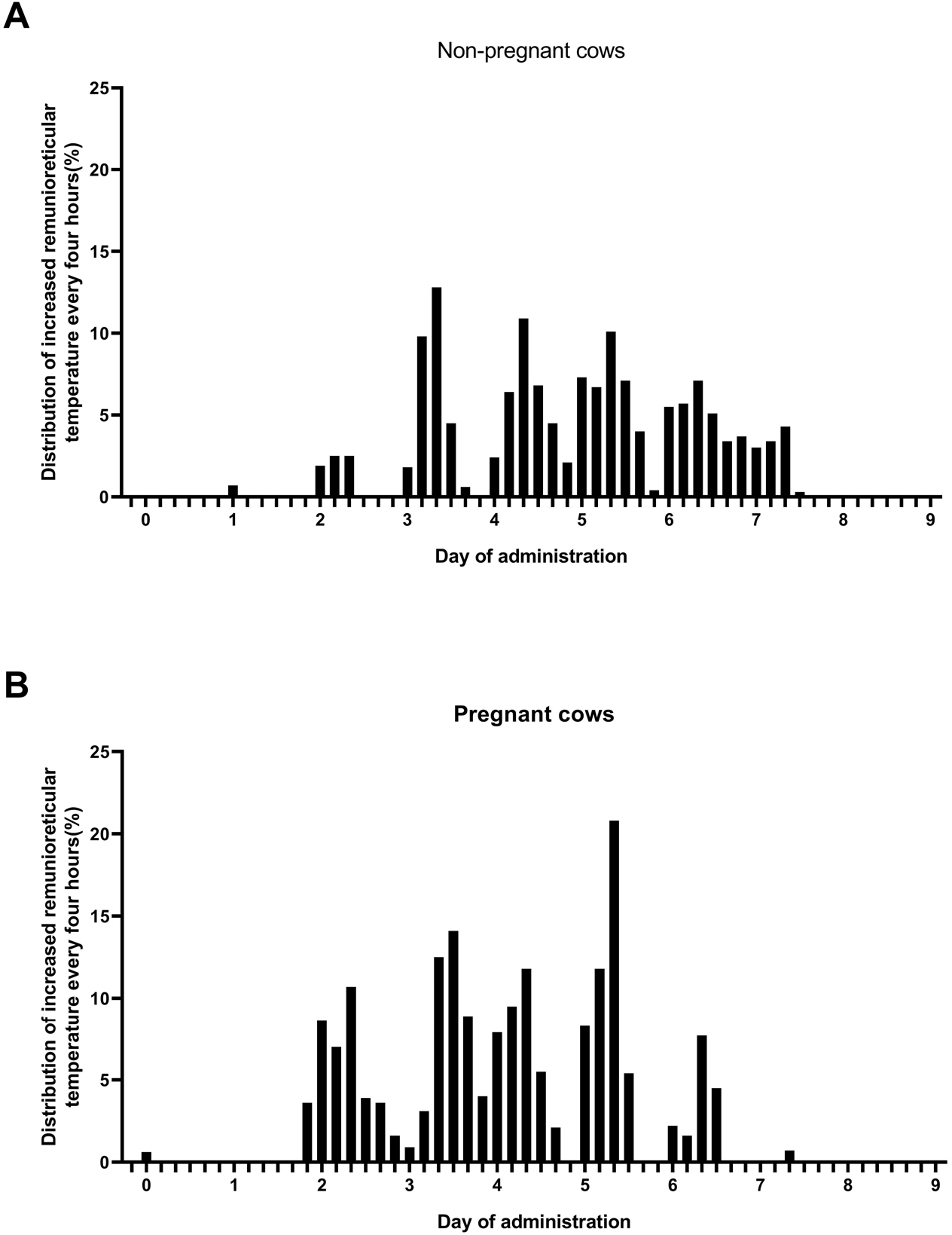
Additionally, the rate of rise in temperature of > 40°C was measured for 4 hours at 10-minute intervals for 9 days after LSD vaccination and analyzed by group. The rate at which a temperature of 40°C was maintained was higher in pregnant cows than it was in non-pregnant cows. A maximum of 12.8% non-pregnant cows and 20.8% pregnant cows demonstrated temperatures > 40°C between 5 and 6 days after vaccination (Fig. 3).
DISCUSSION
There are research reports comparing changes in ruminoreticular temperatures and body activity according to estrus [19], pregnancy [20], parturition [21], and foot-and-mouth disease (FMD) vaccination [22], which are behavioral characteristics of cows using bolus sensors. Because the sensors are located in rumen or reticulum, ruminoreticular temperatures temporarily decreases rapidly due to the effect on water comsumption after ingestion of the feed, and body activity increases than usual because they are mixed with the feed due to feed intake [19,20,23–25]. This characteristic is the result of normal feed intake, so it is also the basis for accurately determining whether cows consume feed.
Governments are encouraging vaccinations to prevent the outbreak of infectious diseases, such as LSD, Akabane disease, and FMD among animals [6,15,22,26,27], considering this is the most efficient way to prevent infection. [6,15,26]. However, the side effects of vaccination must be studied, and vaccination methods to minimize these side effects should be developed [11,16,17,22,23,26].
Recently, Abutarbush et al. [16] reported that LSD vaccination causes fever, decreased feed intake, and reduced milk production in dairy cows. Furthermore, Bamouh et al. [11] found that the rectal temperature increased significantly in the vaccinated group than it did in the non-vaccinated experimental group. Additionally, the body temperature was found to gradually increase up to 6 days after LSD vaccination [11], which conforms to the results of this study. Bamouh et al. [11] also showed that a high vaccine dose caused a rise in temperature to ≥ 40 degrees; this finding was similar to that of our study. Nevertheless, analyzing the changes in temperature depending on the dose of LSD vaccination in cows cannot help elucidate the changes in body temperature according to pregnancy status.
Katsoulos et al. [17] measured rectal temperature using digital thermometers after LSD vaccination and found that the highest rectal temperature was recorded 8 days after vaccination, and milk production decreased by up to 16%. However, analyzing milk production status after parturition also cannot help clarify the changes in body temperature after LSD vaccination according to pregnancy status.
Body temperature is very closely related to physiological mechanisms, and technologies have been developed to monitor body temperature using non-invasive methods, such as by using bolus sensors [22,26]. Our research team previously conducted a study to investigate changes in ruminoreticular temperature and body activity depending on estrus status [19], gestation period [20], and parturition [21]. A study has also been conducted to compare and analyze the ruminoreticular temperature after administering FMD vaccine to cows in early- and late-pregnancy stages [22].
However, to the best of our knowledge, changes in ruminoreticular temperature and activity after administering LSD vaccine during pregnancy have not been analyzed to date. Therefore, the contribution of our study, which shows the relative rise in ruminoreticular temperature of pregnant cows after LSD vaccination when compared with that of non-pregnant cows, is significant.
The current study shows that the rate at which a ruminoreticular temperature of > 40°C was maintained was higher in pregnant cows than it was in non-pregnant cows after LSD vaccination. Hence, prescribing antipyretic drugs and close monitoring are necessary to prevent miscarriage. While no miscarriage or stillbirth occurred while conducting this experiment, additional large-scale studies are required to investigate adverse reactions of LSD vaccination.
In conclusion, the results of this study can be used as raw data to understand the physiological changes in ruminoreticular temperature and body activity depending on pregnancy status after LSD vaccination in Hanwoo. In addition, based on the results of this study, we plan to conduct a study to investigate cases of miscarriage, premature birth, and stillbirth following LSD vaccination in the future and develop ways to prevent them.