INTRODUCTION
Calves were born with an under-developed rumen. Rumen development is accompanied by gut microbial colonization [1]. Feed-induced rumen development has been a key target in calf nutrition, with studies showing an early feeding regime and nutritional strategies can promote early rumen development and the establishment of microbial communities [2]. Additionally, studies indicated that microbial community changes induced through early feeding strategies may persist for 4–5 months [3,4]. Direct feeding of microbes, probiotics, and prebiotics at the weaning period, to manipulate the gut microbiota, have yielded positive results in facilitating the rumen microbial development [5–8]. Thus, neonatal period was considered an opportune window for rumen microbial manipulation [9–11].
The beneficial role of rumen content dosing was realized long before the extensive studies in ruminal microbiome using sequencing technologies. For example, artificial dosing of rumen content of a healthy cow was practiced to treat a sick recipient animal [12], and rumen transfaunation was evaluated for the treatment of indigestion [13] and abomasum displacement in cattle [14]. In early weaned lambs, inoculation of fresh adult rumen fluid into the rumen improved average daily gain and digestibility [15]. In our recently published work, we inoculated the rumen content extracted from an adult cow into the young calves from birth to 8 weeks of age. We observed significant changes in the liver transcriptome along with distinctive microbial communities in the rumen epithelial [16], abomasum [17] and ileum [18] in the calves subjected to early dosing compared to the control animals. Taken together, these studies suggest that artificial dosing of rumen content in neonatal ruminants may offer an effective approach to elicit microbial changes in the gut.
In ruminants, both the rumen and hindgut play significant roles in feed fermentation. However, nutritional studies in cattle primarily focus on the rumen and its microbial environment, leaving the molecular mechanisms and the responses to dietary treatment in the hindgut poorly understood. Previous work indicated that some important vitamins were provided by microbial fermentation in the hindgut, such as vitamin K, thiamine and riboflavin [19,20]. In dairy cattle, hind-gut microbial fermentation is generally responsible for 5% to 10% of total-tract carbohydrate digestion (reviewed in [21]). As part of the hind gut, caecum is the first region of the large intestine, which is a pouch area, where the large and small intestine meet. In lambs, the cecum has a reported role in breaking previously undigested fiber, and producing ~6%–14% of the short-chain fatty acids [22]. In the large bowel and caecum of the cow, the fermentable substrates are limited. These substrates include lignin, crystalline starches unprocessed during foregut digestion and absorption, and some secreted mucins [23]. When dairy cows are fed concentrate-rich diets, large amounts of starch are fermented in the large intestine, leading to hindgut dysbiosis [24,25]. The buffering capacity in the hindgut is limited since it lacks saliva, making the hindgut more susceptible to compromised mucosal permeability and integrity [26].
Like the rumen, microbial colonization in the lower gut occurs after birth. In tangent, the mechanisms governing development and function in the gut change with the microbial community. Few studies investigated the microbial communities in the cecum. Cecal and rumen microbial communities differ in composition and abundance [27]. This difference might be explained by the fermentable substrates in the caecum, which are different from these in the rumen. Godoy-Vitorino and co-authors compared microbial communities of cow cecum and rumen. They reported that the rumen had higher proportions of Bacteroidetes and Spirochaetes, and lower proportions of Firmicutes (Bacillota) and Proteobacteria (Pseudomonadota) compared to the cecum [28]. The cecal microbiota contributes to the post-rumen fermentation of substrates undigested in the rumen. In Holstein steers, cecal fermentation provided up to 8.6% of metabolizable energy intake [29]. Thus, the microbial community changes in the caecum has a perceived impact host’s metabolism. These studies suggested that methods targeting the hindgut might provide new opportunities for nutrient use improvement. However, we still have scarce information on the function of the caecum and its microbial community.
In this study, as a part of a larger study, we focus on investigating the caecum transcriptome changes and its associated microbial communities in calves with or without artificially dosed rumen content extracted from adult cow. Additionally, we compared the microbial communities among the rumen, ileum, abomasum, and caecum tissues collected from the same group of calves using rRNA transcripts generated by RNA-sequencing. This systematic, comparative analysis shed light into the impact of artificial dosing of adult rumen content on microbial communities in various locations of the digestive system in young calves.
MATERIALS AND METHODS
The animal protocols for both the donor cows and young calves approved by University of Wisconsin-Madison, Institutional Animal Care and Use committee (IACUC). All methods were performed in accordance with the relevant guidelines and regulations.
As previously described, two adult cows were used as donor for rumen content preparation as previously identified, they were high milk production efficiency (HE) and low milk production efficiency (LE) cows [30]. Pairwise, HE and LE adult cows were selected using milk production efficiency index (MPI) to avoid the difficulty in comparing animals with drastically different milk production levels. MPI was calculated by dividing energy-corrected milk (ECM) with dry matter intake (DMI) [31]. One adult HE cow (4262; MPI = 1.8, 5 years of age) was selected as the HE donor cow. The pairwise LE cow (4297; MPI = 1.6, 5 years of age) was also identified. The adult cows were offered ad libitum access to water and were fed a total mixed ration (TMR) once daily post-morning milking. TMR ingredient composition was to meet nutritional requirements for lactating dairy cattle established by the National Research Council [32]. On on a dry-matter basis, TMR contained 29.2% neutral detergent fiber (NDF; determined following treatment with sodium sulfite and α-amylase), 43.4% non-fiber carbohydrate, 17.1% crude protein, and 5.0% fat. The detailed TMR information is included in previously published work [30]. This study was performed according to animal protocol A01104, approved by the IACUC of University of Wisconsin-Madison.
For the calves, experiment was performed according to the animal protocol A01501 approved by IACUC of University of Wisconsin-Madison. Eight bull calves were enrolled at birth and randomly assigned into two groups: Treated, inoculated with rumen content from HE donor cow; and control: inoculated with autoclaved rumen content. Besides inoculation, standard herd practices employed by the United States Dairy Forage Research Center farm were carried out throughout the experiment.
The procedure for preparing the dosing inocula was published previously [16]. From the donor cow, fresh ruminal content was removed in the medio-ventral region of the rumen. To make the inoculum, lightly squeezed solids and rumen liquid were mixed by volume in a 3:1 ratio to make the inoculum. The mixture was blended under CO2 immediately for 1 minute, then large particles were removed by squeezing the mixture through four layers of cheesecloth. For treated cohorts, freshly prepared inoculum was used same day. The control inocula were prepared by autoclaving the rumen content. The trial was carried out from birth to 8 weeks. The initial inoculum was administered within 3 days after birth, then at 2, 4, and 6 weeks after that. Calves were euthanized by penetrating captive bolt at 8-week of age and tissue collection immediately followed.
All the calves were subjected to tissue collection. Caecum tissues were collected from each calf immediately after sacrifice. The epithelial layer of the caecum was collected. After being rinsed with 1X phosphate-buffered saline (PBS) to remove any digesta, tissues were cut with sterilized scalpels into small, 4–5 mm2 fragments and put into Eppendorf safe-lock tubes. Collected tissues were snap-frozen in liquid nitrogen, then stored at −80°C until further processing. For microbial community comparative analysis, we included sequence data previously published by our group from these tissue types: epithelial layer of the rumen [16], abomasum [17], ileum [18], and liver [15].
Caecum tissues were first homogenized using Precellys lysing beads CK28 (Bertin Technologies) at 6,800 rpm for 30 seconds for 4 times and the tissue homogenate was stored on ice in between cycles. An miRNeasy kit (Qiagen) was used to extract total RNA samples using a QIAcube (Qiagen) with DNase treatment step added to the protocol. RNA 6000 Nano kit (Agilent Technologies) was used to check the quality of extracted RNA. RNA samples with RNA integrity number ≥ 8.5 were quantified using Qubit 4.0 (Thermo Fisher Scientific) for sequencing library preparation.
Illumina TruSseq ribo-zero gold kit was used for RNA-sequencing library preparation according to manufacturer’s instructions, with 1mg of total RNA as input for each sample. The quality of prepared libraries was analyzed by Bioanalyzer using a DNA 1000 kit (Agilent Technologies). Quantification of library was performed using Kapa quantification kit (Roche, Basel, Switzerland) using a QuantStudio 5 instrument (Applied Biosystems). Initial pooling of the libraries was done using the quantification generated by the Kapa quantification kit by Illumina’s pooling calculator (https://support.illumina.com/help/pooling-calculator/pooling-calculator.htm). The pooled libraries were sequenced using an Illumina MiSeq Nano kit (Illumina). Library pooling was further normalized using the sample index ratio obtained from the Illumina MiSeq Nano kit to ensure equal quantity of eash sample in the pool before final sequencing. Final sequencing of pooled samples was done using an Illumina NextSeq high-output, 300-cycle cartridge on an Illumina NextSeq 500 instrument to generate paired-end, 2x150bp reads.
Minimal group sizes required for RNA sequencing were determined by a power analysis using the Bioconductor Package ssizeRNA [33]. It was determined that 4 biological replicates per treatment with 20 million reads per sample would be sufficient to identify differentially expressed genes (DEGs) from RNA-sequencing data at a power of 0.90 using the following parameters: statistical power cutoff = 0.8, number of genes = 20,000, minimum number of DEGs = 200, average read count = 1,000, fold change = 2, FDR = 0.05. We obtained 50–60 million reads per sample in our experiment design. Thus, the sequencing depth would ensure sufficient statistical power for transcriptome analysis.
For sequence alignment, NCBI ARS-UCD1.3 (https://www.ncbi.nlm.nih.gov/assembly/GCF_002263795.2), Bos taurus reference genome was used. For each sample, raw reads were aligned using STAR [34]. Using gene-level, raw-read counts as the input, DEGs were identified using DESeq2 [35], with p-values corrected for multiple testing using the Benjamini-Hochberg method [36]. To identify confident DEGs, combinatory cutoffs were used to filter DEGs: p-value ≤ 0.05, fold-change ≥ 2 and mean relative change (RC) ≥ 10. Normalized read count, fragments per kilobase of transcript per million reads mapped (FPKM), was calculated using cufflinks [37]. Genes were considered expressed, if the FPKM value is 1 or more. Gene function annotation and gene ontology (GO) analysis were performed using DAVID [38].
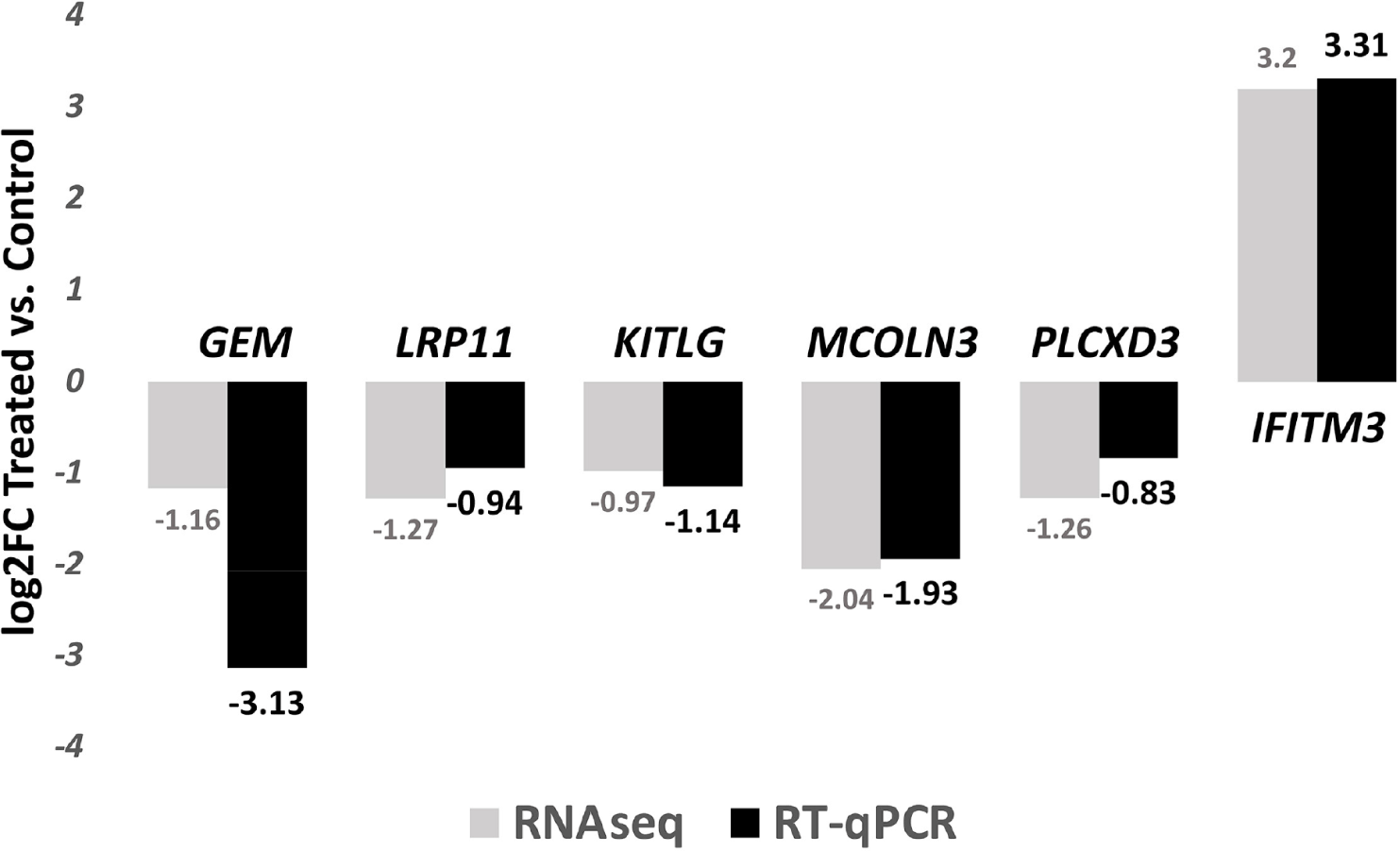
Six randomly selected DEGs identified by RNAseq were analyzed for expression analysis using RT-qPCR: GEM,LRP11, KITLG, MCOLN3, PLCXD3 and IFITM3. GEM encodes a protein that is part of the RAD/GEM family of guanosine triphosphate (GTP)-binding proteins. Associated with the inner face of the plasma membrane, GEM functions as a regulatory protein in receptor-mediated signal transduction [39]. The protein encoded by LRP11 is predicted be an integral component of membrane, and it plays a central role in leptin signaling and regulation of energy homeostasis [40]. KITLG has a reported role in cell migration and proliferation [41]. MCOLN3 protein is a member of the mucolipin family of ion channels, and a novel regulator of trafficking along the endosomal pathway [42,43]. Reduced expression of PLCXD3 is associated with disruption of glucose sensing and insulin signaling in pancreatic β-cells [44]. IFITM3 protein was reported as an important innate immune effector that prevent diverse virus infections in vertebrates [45].
cDNA synthesis was performed using 2 µg of RNA with High-Capacity cDNA master mix (Life Technologies). Gene-specific, Taqman assay probes were ordered from Thermo Fisher Scientific. All real-time qPCR reactions were performed using the QuantStudio 5 (Thermo Fisher Scientific). The qPCR cycling parameter was set as follows: one step of Uracil-DNA glycosylases treatment at 50°C for 2 min, then a denaturation/activation step at 95°C for 2 min, followed by 40 cycles of 95°C for 15s and 60°C for 60s. Triplicate reactions were carried out for each target gene. Gene expression level was normalized to two reference genes, ACTB and HMBS, which were reported with high stability in cattle [46]. The relative quantification of gene expression was determined using the 2−ΔΔCt method [47].
For all the tissue types, rRNA reads from the host microbial community was extracted computationally by the following steps: 1) The total raw reads generated from the RNA-sequencing of the host tissue were first mapped to the genome of Bos taurus (NCBI, ARS-UCD 1.3) using STAR [34]; 2) To enrich rRNA reads, the unmapped reads (non-cattle RNA-seq raw reads) were mapped to rRNA reference using SortMeRNA [48], using the rRNA reference provided by SortMeRNA. 3) The aligned rRNA reads were followed for microbial taxonomic classification, using Kraken [49] (http://ccb.jhu.edu/software/kraken/MANUAL.html). The Kraken database used in the analysis contained ~25,000 complete bacterial, archaeal, and viral genomes in RefSeq.
Genus levels, raw read-counts were used to access the microbial community differences between the treated and control groups using DESeq2 [35], with p-values corrected for multiple testing using the Benjamini-Hochberg method [36]. To identify differentially abundant genera (DAG) with high confidence, the following filtering criteria were applied: fold-change ≥ 2 and p-value < 0.05. Clustering analysis was performed using normalized read-count at genus level with prcomp in R (version 3.2).
To identify the host genes with significant association with its microbes, Spearman’rho analysis was done (SciPy v1.2.0) using the normalized read counts of caecum mRNA and its epimural microbial rRNA. Before the association analysis, we normalized the genus-level read-counts by the following steps 1) Calculating the per-million-factor (PMF) by dividing the total number of reads mapped to genus level by 1,000, 000; 2) The raw read-count at genus level was divided by PMF to get normalized read-count. Only genera with a mean normalized read count less than 5 across all samples were considered for further analysis. DEGs in the caecum were included in the association analysis. To determine statistical significance, these cutoff values were used: p-values ≤ 0.0001 and the correlation coefficient absolute value more than 0.8.
Microbial communities were compared across the rumen, ileum, abomasum, and caecum. For each tissue type, microbial taxa at genus level were included for the analysis. Principal component analysis was performed by including all the tissue types for each treatment group. The top 5% most abundant genera were identified for each tissue type for the treated group, using mean, normalized read count for each genus. Then the shared genera between rumen, ileum, abomasum, and caecum samples were identified. Their abundance in each tissue type was graphed with a stacked bar graph.
RESULTS
On average, a total of 48.1 ± 0.85M reads was obtained for each of the sample. The average mapping rate is 83.11 ± 0.77%. An average of 10,243 ± 341 genes were expressed (FPKM ≥ 1) for each sample. For microbial classification, an average of 2.35 ± 0.17M reads were successfully classified by Kraken. RT-qPCR analysis confirmed the expression profiles as identified by the RNA sequencing method (Fig. 1).
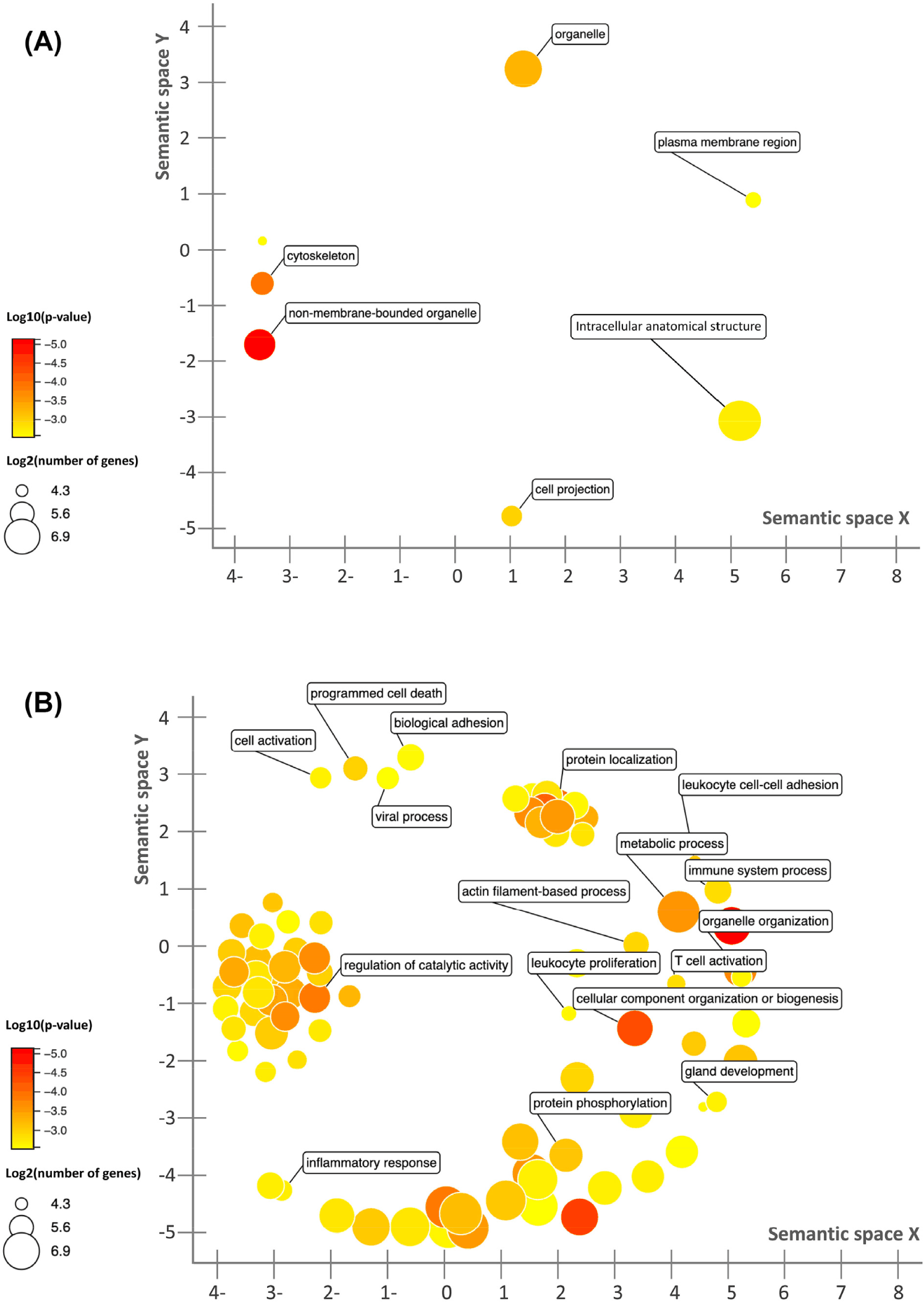
A total of 1,836 genes were identified as significantly differentially expressed (p-value ≤ 0.05, fold-change ≥ 2 and mean RC ≥ 10) between the treated and control groups. Of these, 86 of them had increased expression and 1,750 of them had decreased expression in the treated group compared to the control. GO analysis using the up-regulated genes in the treated group indicated that these genes were enriched in the pathways related to cell proliferation (Fig. 2A). For down-regulated genes in the treated group, a significant enrichment in the immune response, host response to pathogens, and inflammatory responses was observed (Fig. 2B). Ten genes were identified as the top 10 mostly highly expressed genes in the caecum in the treated group, RMRP, SPINK1, EBD, LYSB, OLFM4, CYTB, ACTG2, TPT1, SPINK4, and PIGR. GO analysis indicated that these genes were enriched in the cellular component (GO: 0005576; 5 genes, p-value < 0.001). PIGR encodes a transmembrane protein, which facilitates the transcytosis of the soluble polymeric isoforms of immunoglobulin A and immunoglobulin M in immune complexes [50].
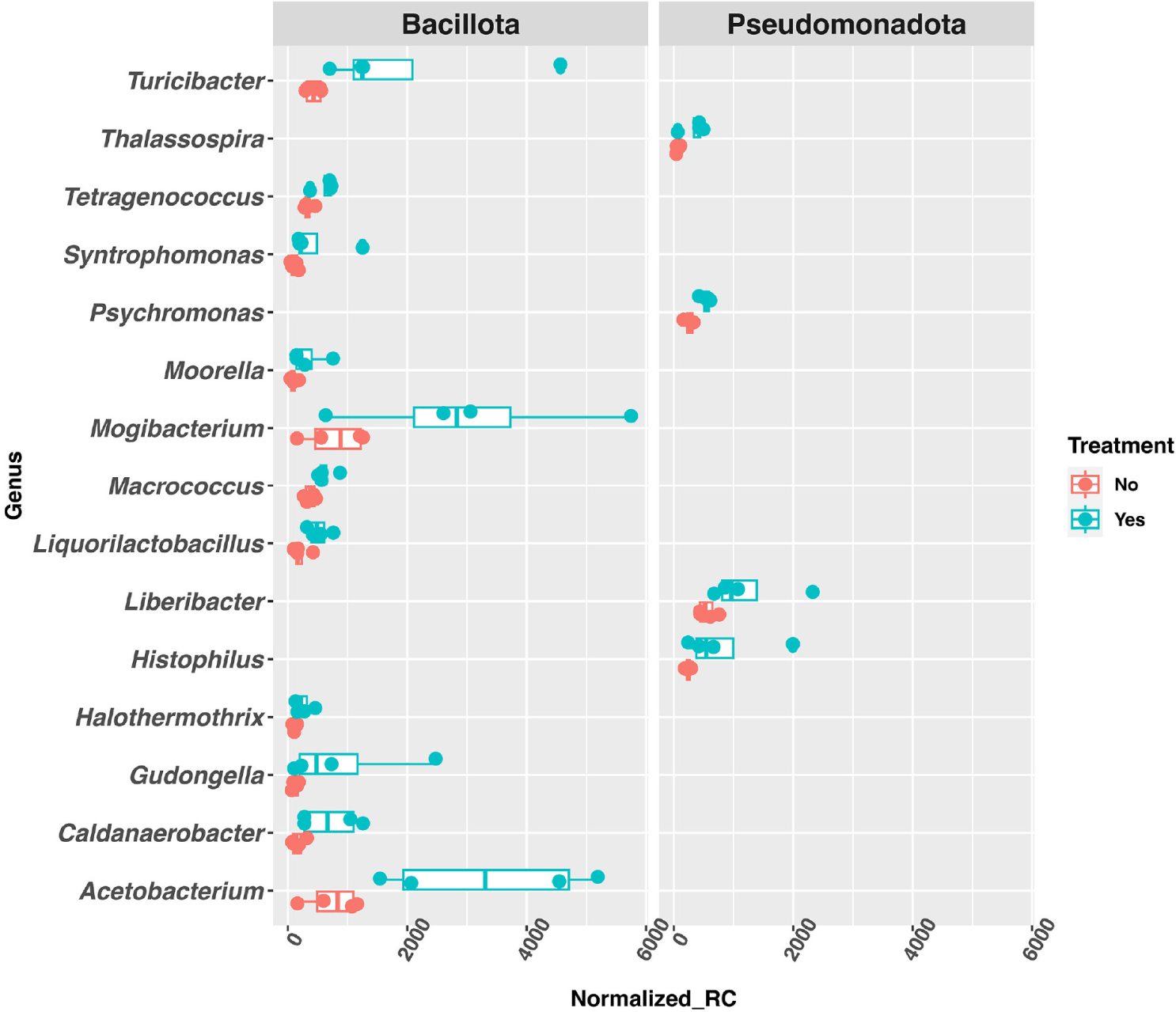
In the caecum, 582 genera were identified with a raw, mean read counts of 5 or more, and 343 genera were identified as DAGs (fold-change ≥ 2 and p-value < 0.05). Among the DAGs, 129 of them showed significant increase in abundance in the treated group, while 214 of them showed significant decrease in the treated group. For the genera with significant abundance increase in the treated group, the top 15 most abundant belonged to two phyla: Bacillota and Pseudomonadota (Fig. 3).
A total of 56 DEGs showed significant association with 20 or more genera in the caecum. Eight of these genes had significant association with more than 40 genera. These genes include RAB26, SLC16A11, RAP1GAP, REEP6, TMEM190, BOLA-DQB, TSPAN32, and CA7. Reactome pathway analysis indicated the enrichment of these genes in the following pathways: Rap1 signaling (BTA-392517, 2 genes, p < 0.01), integrin signaling (BTA-354192, 2 genes, p < 0.01), MHC class II antigen presentation (BTA-2132295, 2 genes, p < 0.02), and adaptive immune system (BTA-1280218, 4 genes, p < 0.01).
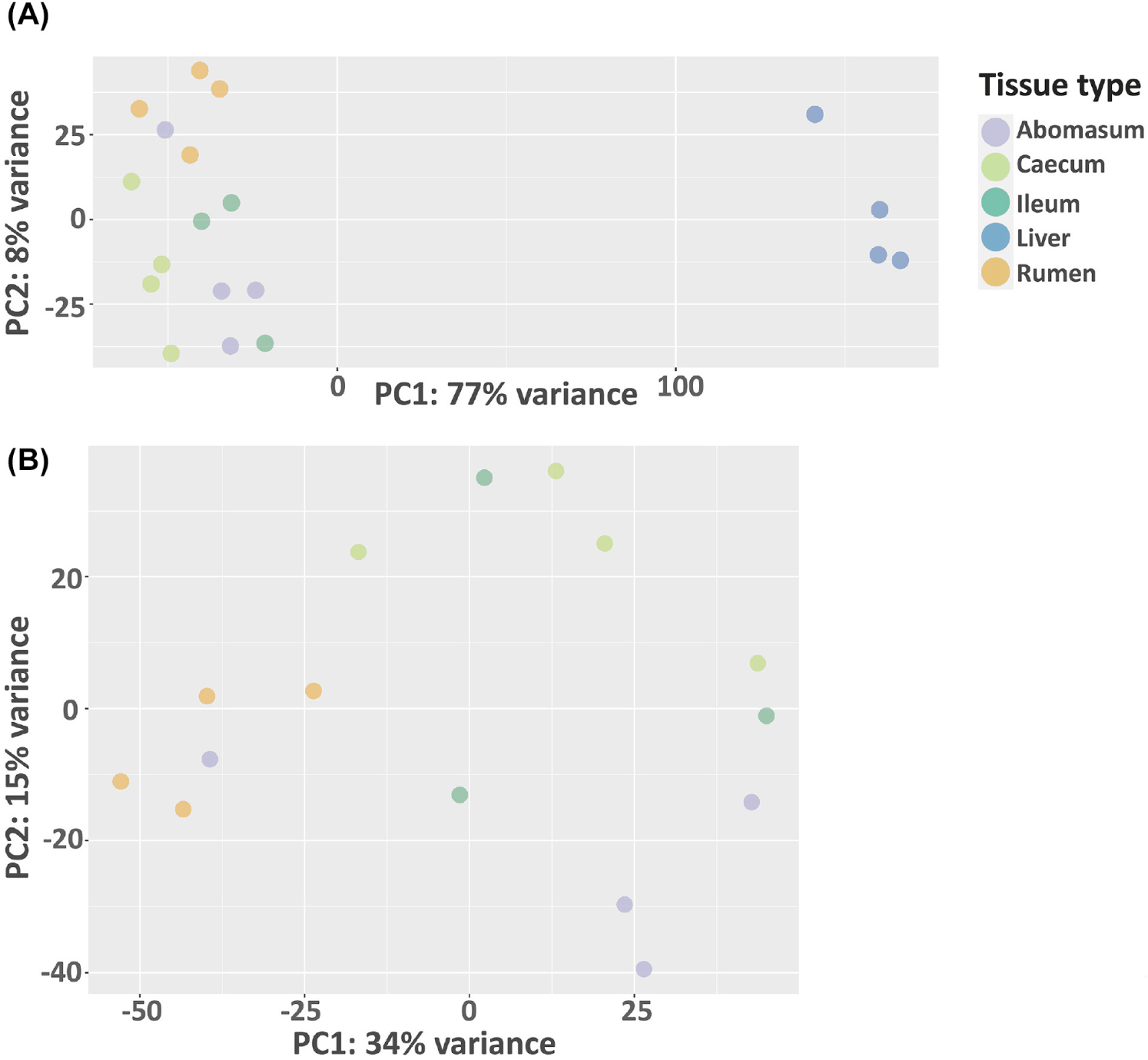
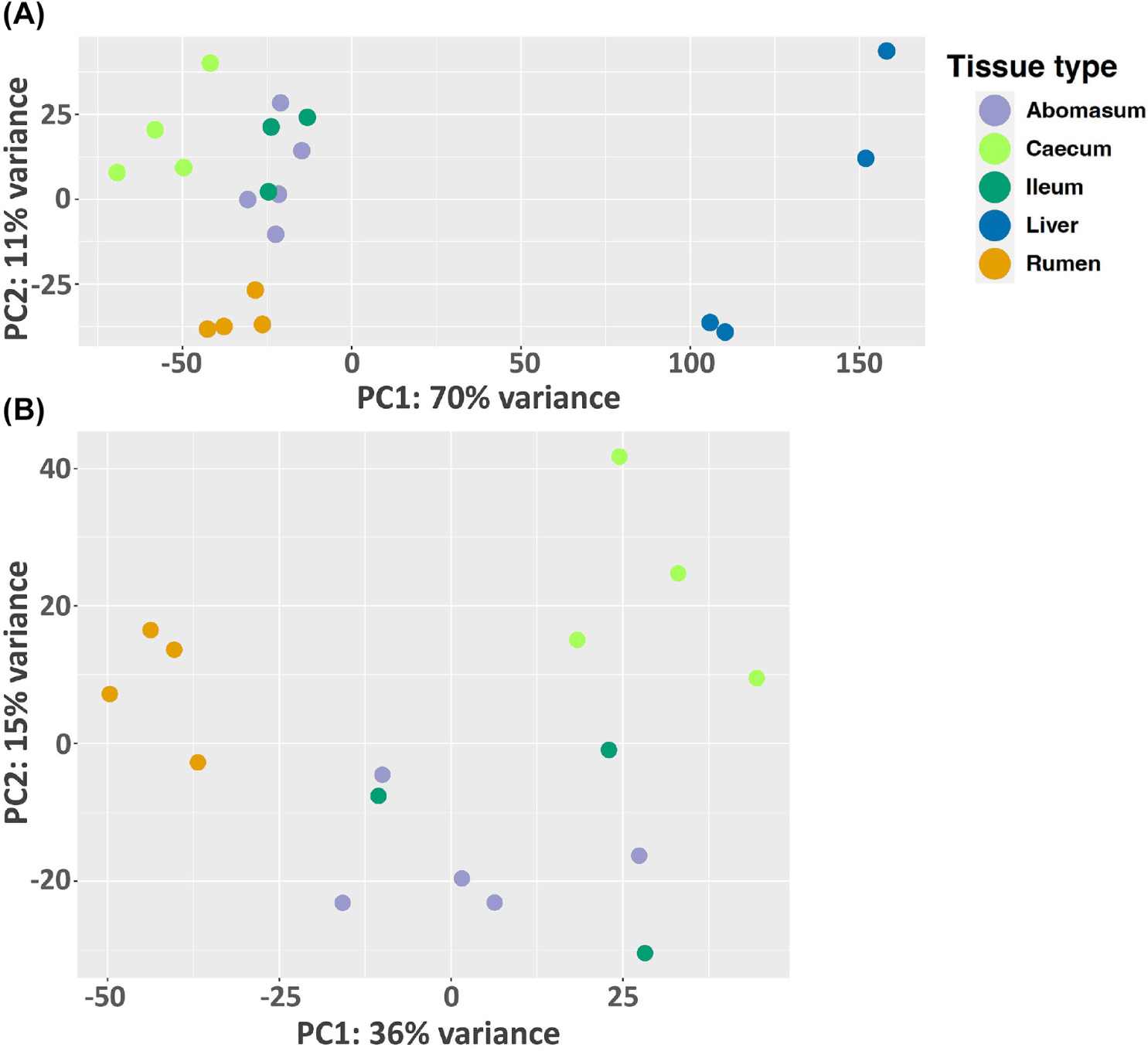
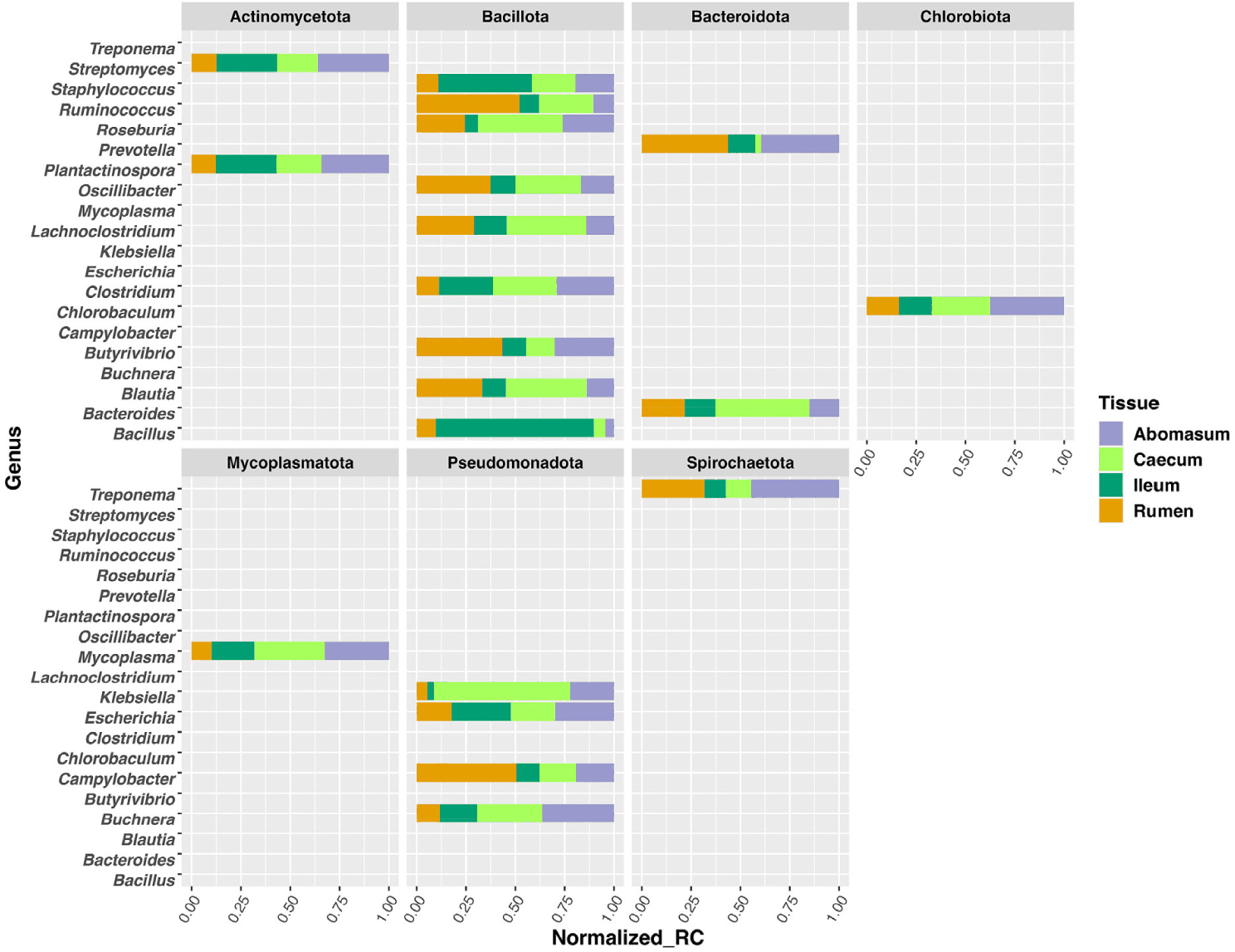
In this study, we analyze the microbial community profile of all the GI tissues we collected from the same set of calves. For both treatments, we observed a clear separation between liver and the other tissues, though the other tissues appear to be mixed under the control condition (Figs. 4A and 4B). For the treated group, the separation of tissue types is clear for the liver, rumen, and caecum. And there is no clear separation between the ileum and abomasum. (Figs. 5A and 5B). Within the treated group, a total of 20 genera, belonging to 7 phyla, were identified as commonly shared amongst rumen, ileum, abomasum, and caecum tissues. However, the abundance of these genera was significantly different (p < 0.05) among these tissues as shown in the stacked bar graph (Fig. 6). The phylum of Bacillota had the highest number of genera shared among these tissue types.
DISCUSSION
For the DEGs associated with the highest number of microbes, GO analysis indicated an enrichment pathway associated with inflammation and immune response. The Rap1 signaling pathway is involved in diverse processes, including cell adhesion, cell-cell junction formation, and cell polarity. Rap1 is a member of the Ras-like small GTPases. It plays a role in the integrin signaling pathway [51], and functions as a regulator of morphogenesis in vivo [52]. Integrins belong to a family of ubiquitous αβ heterodimeric receptors, which exist in multiple conformations and interact with a diverse range of ligands. Additionally, integrins mediate the interactions between cytoskeleton and the extracellular matrix, and they play essential roles in inflammation, infection, and angiogenesis [53–55]. In human studies, it is suggested that the caecum contributes to gut homeostasis and is a major site for generating IgA-secreting cells [56]. Subsequently, secretory IgA cells play a significant role in regulating commensal bacteria populations in animal models [57–59]. The precise role of caecum in host immune system is unknown in cattle. Our findings provided empirical evidence that the microbial communities in the caecum may interact with the host extensively to impact the development of the host’s immune system. Our experiment was done in newborn calves, in which the immune system is immature. Thus, this might be an ideal window to manipulate host immunity and gut microbe colonization through artificial dosing.
As part of the lower gut, caecum is largely ignored in dairy nutrition studies. However, published studies reported significant contributions of the lower gut to host production efficiency [60–62] and immune system maturation [63,64]. In non-ruminants, the interaction between the gut-associated lymphoid tissues (GALT) and gastrointestinal tract (GIT) microbiota affected the functional development of the immune system [65,66]. The stratified keratinized squamous epithelial cells make rumen great for absorption. However, rumen lacks the immunological functionality of the mucosal epithelium present in other regions of the GI tract [67]. GALT is one of the most important immunological tissues since it represents nearly 70% of all the lymphoid tissue [68]. The complete maturation of the GALT tissue is largely dependent on the interaction with GIT microbes [68]. In our study, the downregulated genes in the treated group showed an enrichment in immune response. This finding suggested that the immune response in the epithelial layer of the caecum was impacted by the artificial dosing. Our study only included one type of inoculant with a fixed dosing schedule. The next logical step for follow-up studies will be to investigate if the dosing content and frequency affect or enhance the expression patterns of the immunity related genes in the caecum. The most critical would be to find the core set of microbes that show consistent association with the expression changes in immunity related genes in the host. This set of core microbes may help the development of refined dosing strategy with targeted response.
For the top 10 most highly expressed genes in the treated group, they showed an enrichment in cellular component. This finding indicates that artificial dosing impacted cell structure and proliferation in the caecum. The caecum is a site for generating IgA-secreting cells as reported in human studies. The highly abundant expression of PIGR identified in our study is consistent with the findings in humans [50]. LYSB encodes an intestinal lysozyme with lipolytic activity, which is involved in the disruption of the mycobacteria outer membrane [69,70]. Lysozyme has strong bacteriolytic efficacy and function as defense enzymes against bacterial infections [71]. Thus, the lysozyme is considered an important part of the innate immune system due to its strong antimicrobial activities against bacterial, fungal and viral pathogens [72]. Additionally, in ruminants and animals with foregut fermentation [73], the lysozymes function as a digestion enzyme that degrades the foregut bacteria as a source of amino acids [74]. In our study, the high expression of LYSB in the caecum, a part of the lower gut, might have a dual function in defending the host from the influx of foreign microbes introduced by the artificial dosing, and digesting the excessive number of microbes that are normally not present in calves reared by conventional practice. Consistent with this, Domínguez-Bello et al. reported the activity of bovine gastric lysozyme against pure bacterial cultures [75]. Thus, it’s possible that the high expression of LYSB in the caecum help the host defend against potential infection resultant from the artificial dosing.
Following birth, all regions of the GI tract in the calf undergo microbe acquisition and colonization. Studies have shown that the microbiome diversity and composition differ by region and developmental stage throughout different regions of the GI tract [76–81] and biogeographically (through space and time). We identified 20 genera, belonging to 7 phyla, that are shared between the different GI tissue types investigated in this study. However, the abundance of these genera is significantly different amongst the tissue types included in this study. Thus, it’s evident that the same dosing strategy can lead to differential microbial community in different locations of the gut. This might largely be dependent on the local physiological and ecological environment of the GI tract.
The important role of rumen microbiome in host nutrient use efficiency and production traits has been explored extensively [82–90]. However, studies indicated that feed intake and types have a significant impact shaping ruminal microbiome diversity [89,91,92], making it difficult to assess the exact contribution of rumen microbiome to host production efficiency traits. When investigating the lower gut microbiome and its relationship to host feed efficiency and production traits, Monteiro and co-authors indicated that the lower gut microbiome diversity is less dependent on feed intake and is associated with enhanced ability to digest dietary nutrients. Thus, Monteiro and co-authors suggested that lower gut microorganisms might be correlated with the host milk production traits more than previously appreciated [60]. For future dosing experiments, varied inoculants and dosing schedule may help identify the core set of microbes that persist in the lower gut. And from this group, further analysis that links the host production efficiency and the caecal microbial community can help identify the caecal microbes with a reliable association with host production efficiency traits.