
INTRODUCTION
Cattle production in the Korean Peninsula has a long history, dates back to over 5,000 years [1]. Evidence on the origin and domestication of Korean cattle has been debatable. Genetic evidence based on mtDNA analysis has shown that Korean cattle are maternally traced back to taurine [2]. A genome-wide study using nuclear single nucleotide polymorphism (SNP) markers, however, have detected a substantial indicine introgression in Korean Hanwoo and Japanese Black cattle [3]. Korean indigenous cattle are characterized by different phenotypes and based on coat color phenotypes, which are broadly grouped into four sub-populations or breeds: Brown Hanwoo, Jeju Black, Black cattle (Heugu), and Brindle Hanwoo [1]. Of the four cattle breeds, the Brown Hanwoo is the predominant breed in terms of population size (3,000,000) and geographical distribution, whereas the population sizes of Chikso and Heugu were recently declined [4,5].
The national level genetic improvement program for carcass weight and marbling has been implemented for the Brown Hanwoo cattle for the few decades [1,6]. In line with the measurable changes achieved in growth traits over the last few decades, genome-wide selection scans and association studies of Korean cattle (Hanwoo) did detect putative candidate genes and quantitative trait locus (QTL) associated with carcass weight. For instance, QTL for carcass weight was reported on BTA4 and BTA14 (23–25 Mb) [7–9].
Unlike the Brown Hanwoo, the Black and Brindle cattle breeds have not been targets of strong artificial selection and improvement for specific beef traits; rather a selection against certain phenotypes (e.g., coat color) has led to a steep decline in their effective population sizes. The difference in the breeding programs and population history may have diversely shaped the genomes of Korean native cattle breeds. Assessment of the possible impacts of demographic events on the landscape of genetic diversity and population structure is of particular importance for the ongoing conservation program.
In the absence of phenotypic data, comparisons of breeds that have been subjected to different selective pressures can aid to identify genomic regions and genes controlling qualitative and complex traits. The commercial availability of a wide range of genome-wide SNP panels has been facilitated the fine mapping and detection of genes controlling complex quantitative traits. Previously selection signature scans and genome-wide association studies of Korean cattle have been employed Bovine SNP50 and high-density 777 K chips. In an attempt to identify signatures of divergent selection, we have genotyped three Korean cattle populations showing distinct phenotypes and subjected to different breeding programs using the recently developed GeneSeek Profiler F250 (GGP-F250) array [10].
MATERIALS AND METHODS
Three native Korean cattle breeds Brown Hanwoo (n = 24), Chikso (Brindle Hanwoo, n = 24) and Heugu (Black Hanwoo, n = 24) were sampled and genotyped with the GGP-F250 array, which contains 214677 SNPs. A GGP-F250 Bead Chip was built from diverse Bos taurus and Bos indicus breeds. The SNP markers were screened for the call rate > 90%, minor allele frequency (MAF) > 0.01 and call rate by sample > 90%. Following the application of the above quality control criteria, 108675 autosomal SNP markers were retained and used for downstream analysis.
Genetic diversity was estimated for each breed using PLINK [11]. Inbreeding coefficient and animal relatedness were estimated as the proportion identity-by-decent (IBD) between sample pairs within the breed as a PI_HAT value using the same package. A principal component analysis (PCA) was also performed on 108,675 SNPs using SNP and Variation Suite v8.5.0 (Golden Helix, Bozeman, MT, www.goldenhelix.com). Linkage disequilibrium (LD) was estimated between adjacent SNPs. Inter-SNP distances were binned into different categories and estimates of r2 were averaged for each inter-SNP distance category. Effective population size (Ne) in each breed was estimated from linkage disequilibrium using SNEP tool [12] with the default settings. Based on the population structure and PCA results, FST values were calculated between Hanwoo-Chikso, Hanwoo-Heugu, and Chikso-Heugu according to [13] in sliding windows of 20 SNPs. Windows of the SNP were used to minimize the stochastic effect of single SNP. Fst values were converted to a standard normal distribution (ZFst). The SNP windows that display high genetic differentiations (ZFst ≥ 5) were considered as candidates. Candidate genes within the top ZFst values were annotated using the Bovine UMD.3.1 genome assembly. QTL regions that overlapped with the identified candidate regions were searched from the cattle QTLdb (http://www.animalgenome.org/cgi-bin/QTLdb/BT/index).
RESULTS AND DISCUSSION
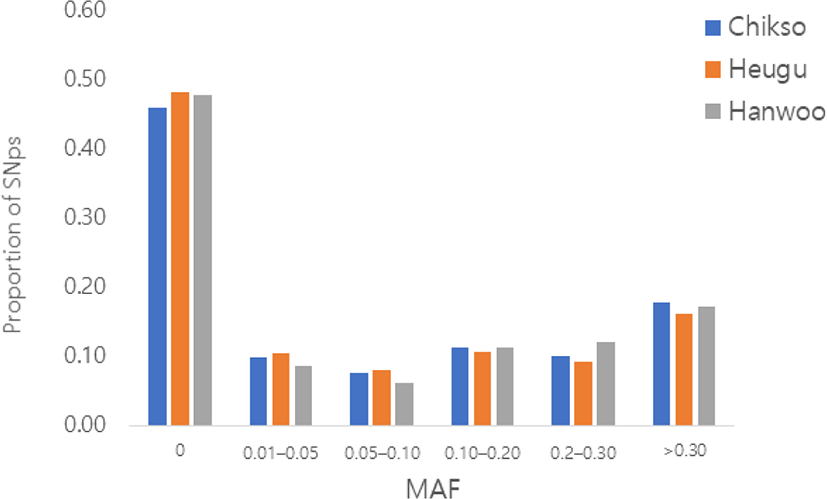
Of the total autosomal SNPs (191,789), 47% were found to be fixed across the three breeds. The percentage of SNPs with MAF > 5% was 44%, 41%, and 44% in Hanwoo, Heugu, and Chikso, respectively. The proportion of polymorphic SNPs observed in the Korean cattle is nearly similar to the figure observed on GGP-F250 (https://genomics.neogen.com/pdf/slicks/ag229_ggp_f250_array_flyer.pdf) for diverse cattle breeds (Fig. 1). This higher proportion of fixed SNPs in our study cattle breed could be explained by the fact that the GGPF-250 array largely contained functional variants (non-synonymous, frameshift and premature stop) having a functional effect or causative polymorphism and subject to selection. Previous studies have shown that functional SNPs (non-synonymous) had lower minor allele frequencies [14,15]. The SNP was considered as breed-specific when it was polymorphic (MAF > 0.05) in the particular breed, but fixed in the other breeds [16]. The highest number of breed-specific SNPs (1784) was found in the Korean black cattle, while the Hanwoo breed had a lower number of breeds specific SNPs (925). The average MAF of breed-specific SNPs ranged from 0.09 in Chikso to 0.14 in Heugu. The SNPs identified as specific to one breed can be used for product traceability and breed discrimination/assignment.
To assess the consequences of artificial selection and demographic events in shaping the genetic constitution of Korean native cattle breeds, level of within breed genetic variability was assessed based on observed and expected heterozygosity. Results show that a deficiency of heterozygosity was detected in the three cattle populations. Specifically, the Korean black (Heugu) cattle retained the lowest level of within-breed genetic variation (Table 1). The observed heterozygosity in Hanwoo and Chikso are higher than the values (0.26–0.29) estimates from high-density SNPs [17], but far lower than the microsatellite (0.67) [5] and Bovine SNP50 BeadChip based estimates (0.38–0.44) [18]. The low level of genetic variability obtained from Heugu is possibly attributed to genetic drift associated with low effective population and geographical isolation [4]. The estimated inbreeding coefficients were –0.041, –0.069, and –0.045 in Chikso, Heugu, and Hanwoo, respectively. The estimated average relatedness was highest for Heigu (0.155) followed by Hanwoo (0.12).
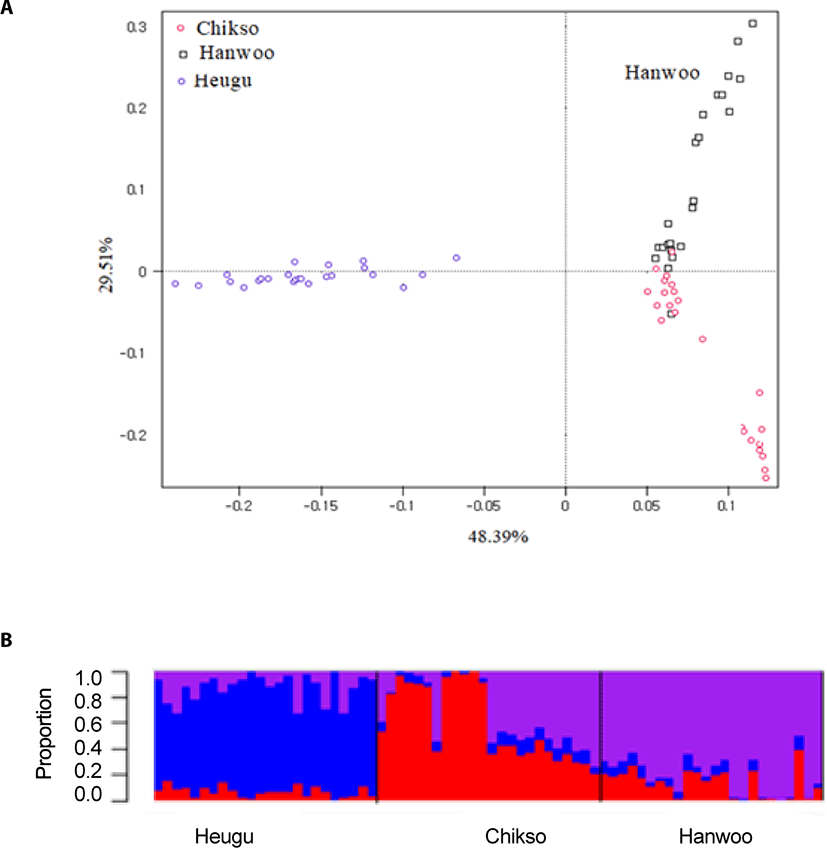
To portray population structure, a principal component was carried out and results showed that the three breeds were clustered based on their coat color phenotypes and breeding histories. As well depicted in Fig. 2, the PC1 accounted for about 48.39% of the variation and prominently separated Hanwoo and Chikso from the Korean Black cattle with a lower level of variation in Heugu. The PC2 segregated Brown Hanwoo population from the Brindle Hanwoo (Chikso) with some individuals from both breeds clustered closely (Fig. 3). In addition, Heugu breed was far separated from the two mainland cattle breeds, possibly due to geographical isolation and genetic drift associated with small effective population size. The PCA results further supported by a model-based population structure (Fig. 2).
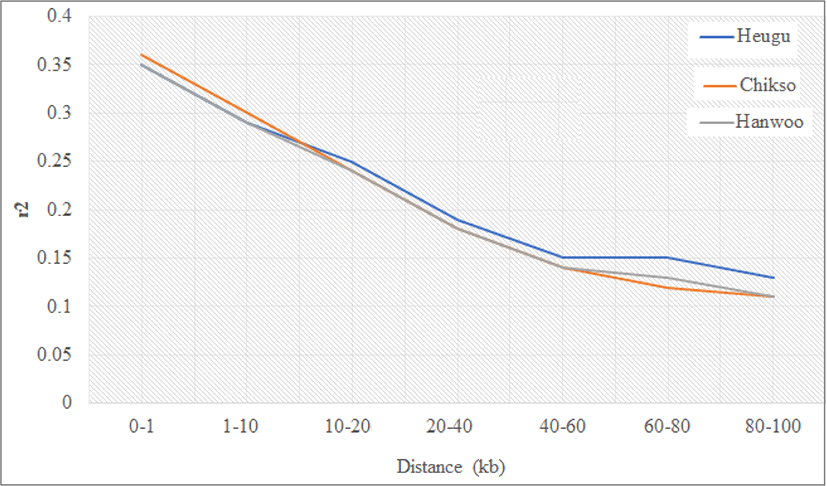
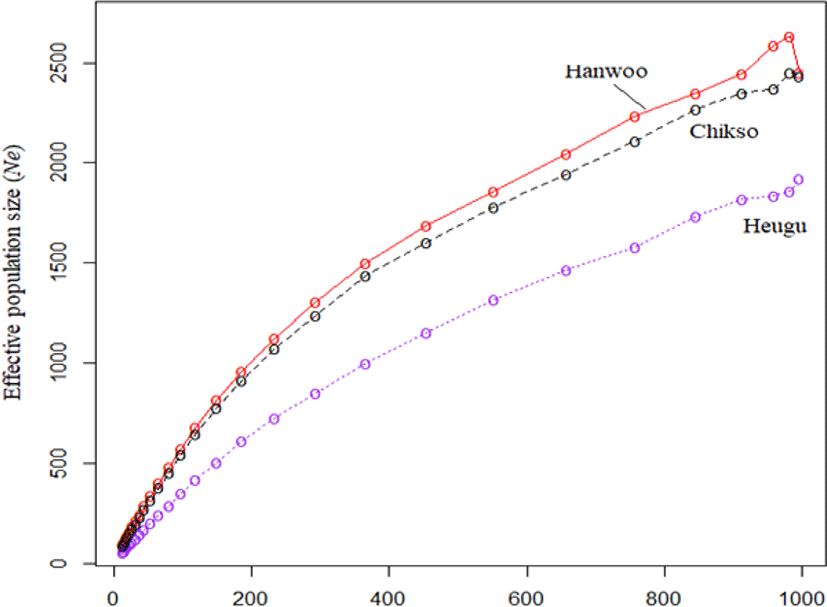
Linkage disequilibrium as estimated by r2 were found to be 0.25 ± 0.34, 0.26 ± 0.35 and 0.26 ± 0.35 in Hanwoo, Chikso, and Heugu, respectively. These estimates were higher than the reported 0.1, 0.13, and 0.12 values in Hanwoo, Chikso, and Heugu based on 50 K chip analysis [18]. Across the three breeds, r2 values decreased steeply as the distance between markers increased. For instance, in the Hanwoo cattle, r2 values dropped from 0.35 to 0.11 when the inter-marker distance increased from 0–10 to 80–100 b (Fig. 4). The higher LD at a shorter distance in the three Korean cattle breeds could be attributed to a reduction in their effective population sizes across generations. The effective population size (Ne) during the last 13 generations (~65 years) in Hanwoo, Chikso, and Heugu were 88, 94, and 57, respectively. Overall, the studied breeds showed a consistent decline in Ne across all generations. Of the three Korean native cattle breeds, Heugu had smaller Ne values at all generations considered (Fig. 5). These smaller Ne values obtained from Heugu are in concurrent with the estimated lower within-breed genetic diversity. The Ne values obtained in this study for the last 13 generations are far lower than those reported for Hanwoo (260) and Chikso (202) [17], but higher than the values estimated for Hanwoo (83) and Chikso (59) [18]. This difference can be attributed to the difference in genotyping arrays, Bovine HD and SNP50, respectively.
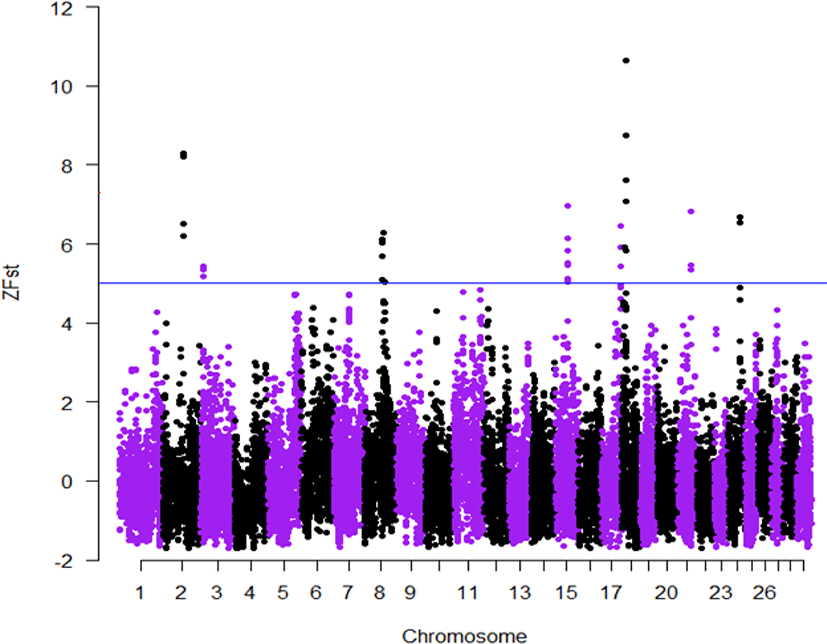
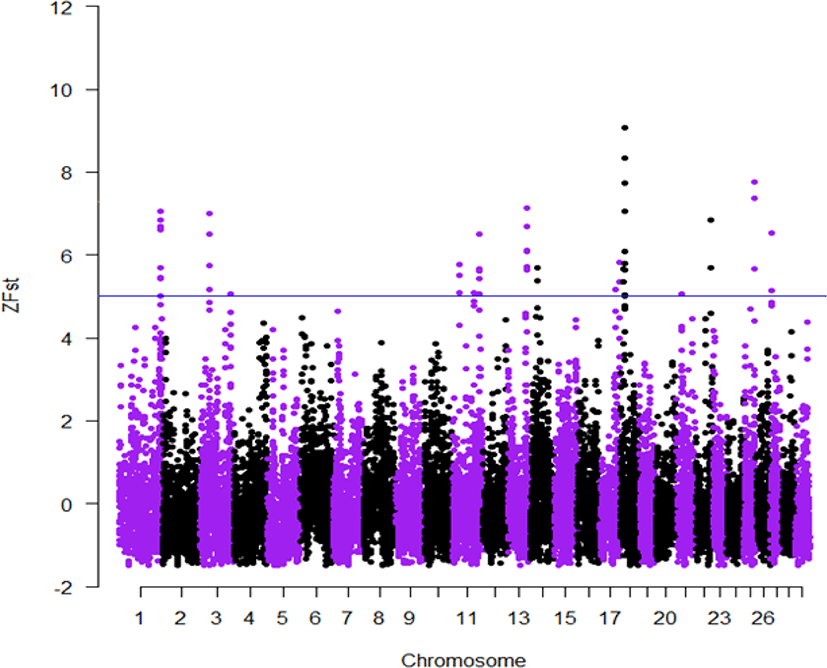
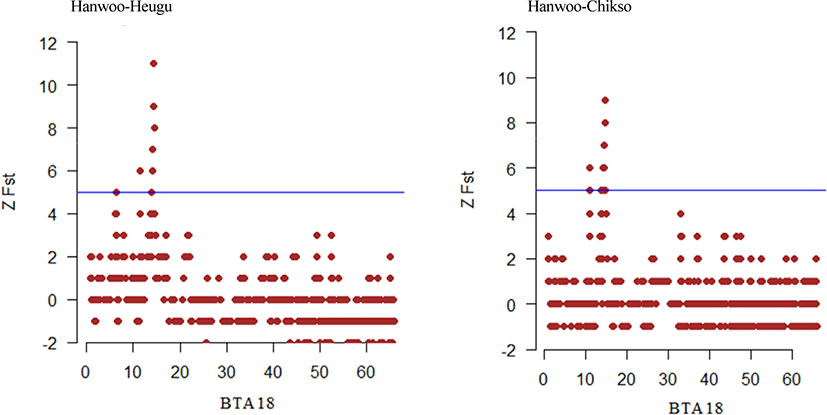
Here for the first time, we employed the GGP-F250 chip primarily built from functional variants to investigate how selection and genetic drift have shaped the genetic variation of Korean native cattle breeds displaying distinct phenotypic traits. Tables 2 and 3, present candidate regions and genes identified in the comparisons of Hanwoo-Heugu and Hanwoo-Chikso, respectively. Across the bovine autosomal chromosomes, 113 and 84 genes were detected for Hanwoo-Chikso and Hanwoo-Heugu pairs analyses, respectively. For the two pairs of comparisons, the strongest signals of selection were detected on BTA18 (12.51–14.57 Mb; ZFst = 11). The largest numbers of candidate genes were also located on BTA18 followed on BTA8 and BTA11. Specifically, the region on BTA18 between 13.34 and 15.06 Mb includes 25 genes in Hanwoo-Chikso pair. For Heugu-Hanwoo group, the candidate regions on BTA18 (13.93–14.68 Mb) contained 18 genes. The strong signals of selection on BTA18 also had strong linkage disequilibrium and diverse haplotypes in the three cattle breeds analysed (Figs. 6–8). In addition to variation in production traits, the studied breeds or breeds under study also exhibit distinct coat colors and patterns. The Heugu cattle display a solid black coat color, whereas Hanwoo shows brown coat color. Interestingly, the region on BTA18 (14.32–14.68 Mb; ZFst = 8) showing strong divergent selection signals was only 19 kb away from the MC1R gene (14,705,093–14,706,843 [Figs. 6–8]).
As compared with the two Korean native cattle breeds, the Brown Hanwoo cattle breed has been phenotypically subjected to selection for beef production and quality traits, including carcass weight, eye muscle area, backfat thickness, and marbling score for the past 30 years [6]. The list of our candidate genes includes genes affecting body size, growth, stature, and carcass quality traits (Table 2 and 3). More importantly, the region between 24.67 Mb and 25.65 Mb on BTA 14 includes XKR4, PLAG1, LYN, DPEP1, SDR16C5, and TMEM68 genes that have been previously well known to affect several traits including height, carcass weight, and stature. This region also overlapped with several known QTL for stature, yearling weight, weaning weight, body weight gain, subcutaneous rump fat thickness, intramuscular fat, average daily gain, and carcass weight. The XKR4 gene has been reported to be associated with the backfat thickness [19,20], feed intake, and average daily gain [21,22]. PLAG1 and LYN gene were associated with variation in stature in cattle and humans [23,24], associated with feed intake and growth in beef cattle [25]. TMEM68 is associated with several production traits including growth, birth weight, feed intake, meat tenderness, and growth/yearling weight in cattle [22,26]. SDR16C5 is associated with birth weight, body weight, and differences in body stature in beef and dairy cattle breeds [24,27]. Additionally, this gene influences human height [23,28,29]. In agreement with our results, a genome-wide association study of Korean cattle (Hanwoo) did detect QTL for carcass weight on BTA14 at 23–25 Mb [8,9].
We further assessed selective signals between the two pairwise comparisons. Common signals of selection between Hanwoo-Heugu and Hanwoo-Chikso pairs were identified on 3 autosomal chromosomes including on BTA11 (93.79–93.80 Mb), BTA17(73.00–73.02 Mb), and BTA18 (14.06–14.68 Mb). Of these candidate regions, the strongest shared selection signals were observed on BTA18 (13.34–15.07 Mb) that covered 1,727 kb. This region includes 18 genes (ACSF3, ANKRD11, APRT, CBFA2T3, CDH15, CDT1, CPNE7, FANCA, GALNS, IGLL1, OR1L1, OR1L3, RF00026, SLC22A31, SPG7, TRAPPC2L, ZNF276, ZNF280A [Table 3; Figs. 6 and 7]). The ACSF3 gene has been identified as a novel candidate for intramuscular fat (IMF) [30] and gene activates fatty acids [31]. Two of the candidate genes (CDH15 and GALNS) have reported to be associated with meat production and stature in cattle [32]. Our LD and haplotype analysis did detect strong LD and haplotype block in the GALNS, CDH15, and APRT gene regions. In their expression analysis, [33] demonstrated that the expression of DPEP1 gene was increased in cattle with greater average daily gain. CPNE7, FANCA, and SPG7 all located on BTA18 between 13.93 and 14.68 Mb were identified as being under selection in European taurine breeds and linked to reproduction, lipid metabolism process, and nervous system development [34]. These shared signals of selection in genes associated with beef traits could imply that Hanwoo cattle are selected for growth and carcass quality traits.
Several earlier and recent studies have clearly shown that growth hormone (GH) plays an important role in controlling growth rate, feed intake, lactation, and carcass traits [35–37]. GH levels are regulated by the release of GHRH [38]. Coincidentally, a region on BTA13 (66.66–66.86 Mb) spanning 5 genes, including RBL1, SOGA1, MROH8, RPN2, and GHRH. Mutations in this GHRH gene are linked to carcass yield in cattle [39]. Previous studies have shown that SOGA1 gene affects average daily gain [40,41] and residual feed intake [40]. A region spanning 4.65–5.04 Mb on BTA27 harbored candidate gene for abiogenesis and fatty acid oxidation [42]. Another potential region of interest is flagged on BTA22 (52.18–52.76 Mb) containing the CDC25A which underlines body size variation and under strong selection in beef cattle [43]. Another candidate region is BTA5:100.36–101.29 Mb, which spanned the CLEC12B gene. The CLEC family genes such as CLEC12A and CLEC4G are highly expressed in tender meat and associated with meat tenderness [44,45]. Another gene also within the BTA17 (54.27–54.40 Mb), EIF2B1, was identified as a potential candidate for beef tenderness [46]. BANP is associated with preadipocyte proliferation [47]. Following the association of several of our candidate genes with meat quality traits, further functional analysis of the divergently selected genes (DBI, ACSF3, HINT2, GBA2, AGPAT5, SCAP, ELP6, APOB, and RBL1) was associated with relevant gene ontology (GO) terms including fatty oxidation, metabolism, biosynthesis, and storage.
The expression of DPEP1 gene was increased in cattle with greater average daily gain [33], expression of GALM is lower in cattle showing lower IMF [33,48]. Several of the candidate genes found in this study were previously reported QTL regions on beef production traits (Tables 2 and 3).
In summary, based on the results of the present study, the genetic diversity and population structure of Korean cattle breeds can be revealed by population demographic events and selective breeding. The smallest effective population size obtained in particular for Heugu over generations pledges further strengthening of the ongoing conservation program. The candidate genes detected in the present study are previously known to affect beef production and quality traits and could be used in the beef cattle breeding strategies.