Treponema phagedenis is a fastidious anaerobe recognized as a major bacterium in bovine digital dermatitis (BDD) [1], a polytreponemal foot disease that causes lameness in cattle due to painful lesions and has serious effects on both animal welfare and economic efficiency [2]. The persistence of T. phagedenis in the interface between affected and healthy tissues [3] and its frequent detection and abundance in BDD suggests that it is a key agent in BDD development [4,5]. The pathogenicity of T. phagedenis is unclear, suggesting the need for further investigation because only a limited number of studies are available. This study reports the whole genome of T. phagedenis KS1, which should assist in research aimed at identifying putative pathogenicity related factors in T. phagedenis.
T. phagedenis KS1, the first BDD-associated Treponema spp. strain identified in Korea, was isolated from cattle with BDD [6]. Biopsy material from a BDD lesion was cleaned, inoculated to oral Treponema enrichment broth (OTEB, AnaerobeSystems, Morgan Hill, CA, USA) containing 10% fetal bovine serum, rifampicin, and enrofloxacin (10 µg/mL), and then incubated anaerobically at 37°C for 7 days. Purity was obtained by subculturing on fastidious anaerobe agar with 5% sheep blood and supplementations and growth conditions as above. The strain exhibited weak β-hemolysis, agar penetration due to its motile and chemotactic nature, and microscopic round body formation, which are rarely reported for other T. phagedenis strains and are considered putative virulence factors. The strain also exhibited increased resistance toward enrofloxacin and kanamycin. QIAamp DNA kit (Qiagen, Germantown, MD, USA) was used for DNA isolation performed by following the standard protocol. The DNA sample was submitted to Macrogen (Seoul, Korea) for sequencing with DNA condition and quantity pre-assessed. Libraries were constructed using the 20 kb SMRTbell and TruSeqNano DNA Kit for the PacBio RSII and Illumina HiSeqXTen platforms, respectively, and the results were quality checked using Qubit and Picogreen. The filtered RSII and HiSeqXTen raw reads produced 133,060 single-pass reads and 6,909,284 high-quality short-reads, respectively. De-novo assembly was done by mapping the PacBio RSII single-pass reads to seed reads using six assemblers and were compared based on total number of bases, N50, number and minimum and maximum lengths of contigs. Assembly by Hierarchical Genome Assembly Process (HGAP3) was selected based on the mentioned criteria. Subsequently, the filtered Illumina high-quality short-reads were assembled to the RSII scaffolds for sequence compensation and error correction using Pilon v1.21 [7], thereby constructing the contigs more accurately. By mapping the Illumina subreads against the PacBio assembled contigs, the consensus sequence with depth of coverage data was generated. Post-assembly validations were conducted, including K-mer analysis, reads mapping, BLAST analysis, and BUSCO analysis. Homology with database sequences was analyzed to infer the structural annotations derived by using Prokka v1.12b [9]. Functional annotation based on ortholog identification was carried out by using EggNOG v5.0 [10].
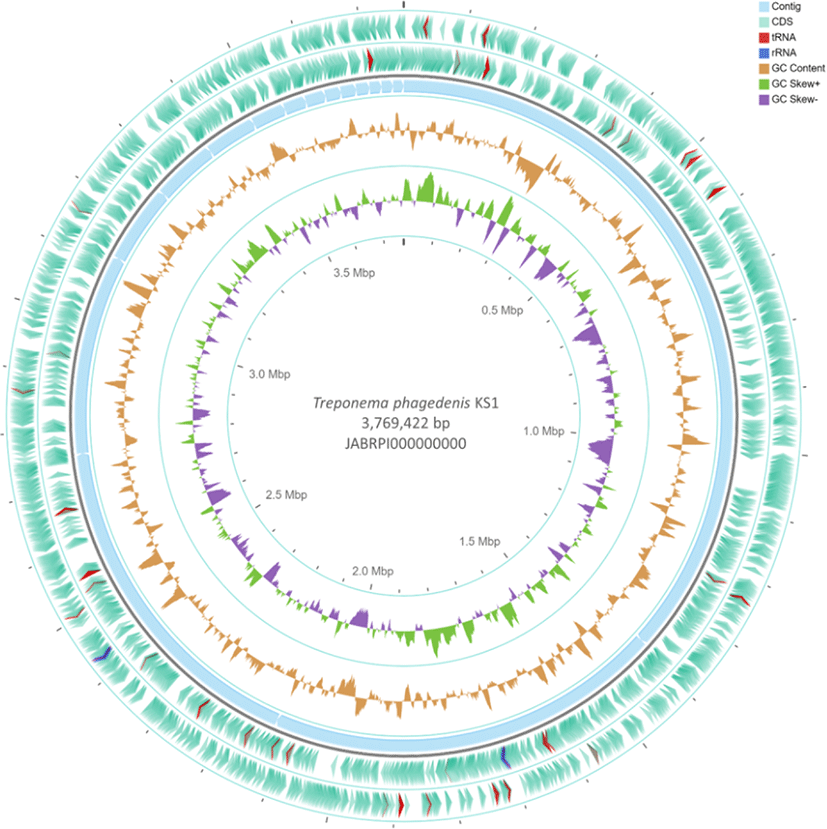
The general properties and functional annotation of T. phagedenis KS1 draft genome were summarized in Table 1 and the circular genome representation was illustrated in Fig. 1. Several spirochetes are classified as pathogenic because of their capability to invade a wide range of tissues in its host. The distinct shape and motility of spirochetes modulated by its periplasmic flagellum allows the bacterium to move through tissues making it to be highly invasive in mammals [11]. Out of 3,364 coding sequence (CDS) categorized, 1.3% are under cell motility, and 1.9% were categorized under signal transduction mechanisms. These included several methyl-accepting chemotaxis proteins (Mcp’s), chemotaxis proteins (Che’s), flagellar motor switch proteins (fli’s), and flagellar protein (Fla’s), and motility protein (motB) for cell motility and signal transduction mechanisms, respectively. Also, 2.26% of the CDS were categorized under defense mechanism which included multidrug resistance protein (norM), multiple antibiotic resistance (MarC)-related protein, methicillin resistance (femX), Beta-lactamases (flp) and desiccation/radiation resistance gene. Other putative pathogenicity-related factors predicted include one CDS encoding hemolysin III family protein, as well as three CDS encoding hypothetical proteins for Mu-like prophage (Flumu protein gp29) and 58 hypothetical proteins for transposases. A total of eight hypothetical proteins encoding for surface antigens with unknown functions, and three genes (tpf1, smc1, and tpd) encoding for surface antigenic proteins were also predicted. This report provided useful insights on the putative pathogenicity-related factors of T. phagedenis KS1, as well as its antigenic components which could be used for diagnosis and prevention, and treatment strategies of BDD.