
INTRODUCTION
The thiazolidinediones (TZD) family specifically bind to and activate proliferator-activated receptor-γ (PPARγ), increase its expression and that of the related genes (e.g., CCAAT/enhancer-binding protein alpha [C/EBPα]), and promote adipocyte differentiation. Yeow et al. [1] demonstrated that the TZD rosiglitazone, in the absence of supplemental fatty acids, strongly up-regulated PPARγ expression in C2C12 cells, and proposed that rosiglitazone inhibited myogenesis. Singh et al. [2] reported that the TZD ciglitazone up-regulated PPARγ and C/EBPα gene expression and depressed myoblast fusion in porcine satellite cells. In our previous study, it was reported that bovine muscle satellite cells (BSC) cultured in a growth medium supplemented with oleic acid (18:1n-9) increases the PPARγ and C/EBPβ gene expressions in myotubes regardless of whether ciglitazone is added or not [3]. Teboul et al. [4] first reported the almost complete conversion of C2C12 myoblasts to lipid-filled cells when incubated with linoleic acid (18:2n-6) plus the TZD pioglitazone, as indicated by a cessation of myoblast fusion, swelling of myoblasts through lipid filling, down-regulation of myogenin (MYOG), and up-regulation of adipocyte lipid-binding protein gene expression. In our previous study [3], we established that 10 µM ciglitizone down-regulated the expression of genes associated with myogenesis, and up-regulated genes involved in adipogenesis. However, we did not establish that 10 µM ciglitizone would elicit maximal effects on adipogenesis and/or myogenesis. Therefore, we hypothesized that a greater concentration of ciglitazone would more strongly promote the adipocyte phenotype in bovine BSC. For the current study, we selected a differentiation medium containing Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 2% horse serum based on the study of Singh et al. [2], which described the transdifferentiation of porcine satellite cells into adipoblasts. Here, however, we altered the adipogenic mixture of that study such as the DMEM, horse serum, penicillin/streptomycin, ascorbic acid, biotin, acetic acid, pantothenic acid, dexamethasone, isobutyl ethylxanthine, insulin, and ciglitazone to include increasing concentrations of ciglitazone (5, 10, and 20 µM). We changed the traditional cocktail of differentiation and studied the effect of ciglitazone alone on adipogenic transdifferentiation of BSC without other factors promoting lipid deposition. RNA-seq was used to further analyze and validate the changes of adipogenesis-related genes and pathways after treatment with different concentrations of ciglitazone. We added horse serum to the medium to promote BSC proliferation and myogenic gene expression to better reflect the in situ conditions.
MATERIAL AND METHODS
All experimental procedures involving animals were approved by the Institutional Animal Care and Use Committee of Yanbian University using the approval code YBU-20160303.
Using sterile techniques, the semimembranosus muscle was collected immediately after the slaughter of Yanbian yellow cattle. The connective tissues and visible fats were removed, and the muscle tissues were ground using a sterile meat grinder and incubated with 1% Pronase (10165921001, Roche, Basel, Switzerland) in EBSS (Sigma-Aldrich, Louis, MO, USA) solution at 37°C for 1h [5]. Following the enzymatic digestion was the repeated centrifugation according to the procedure previously published [3]. The obtained primary cells were suspended in DMEM (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) containing 10% fetal bovine serum (FBS) (Gibco, Thermo Fisher Scientific) and 10% (vol/vol) dimethyl sulfoxide (Sigma-Aldrich), frozen, and stored in liquid nitrogen for subsequent experiments. The BSCs were cultured in DMEM containing 10% FBS, and 1× antibiotic-antimycotic (Gibco) in a humidified incubator at 37°C with 5% CO2. The growth medium was replaced with a differentiation medium after the cells have reached 80% confluence. The differentiation medium of Singh et al. [2] was followed omitting the insulin, pantothenic acid, and dexamethasone, and supplemented with 5 µM ciglitazone (dissolved in ethanol, Sigma-Aldrich) (CL), 10 µM ciglitazone (CM), or 20 µM ciglitazone (CH). Each experiment was replicated in three independent cultures (passage no. 2 of the BSCs were used). The average cell size and viability were determined using the Luna automatic cell counter (Luna II, Logo Biosystems, Anyang, Korea).
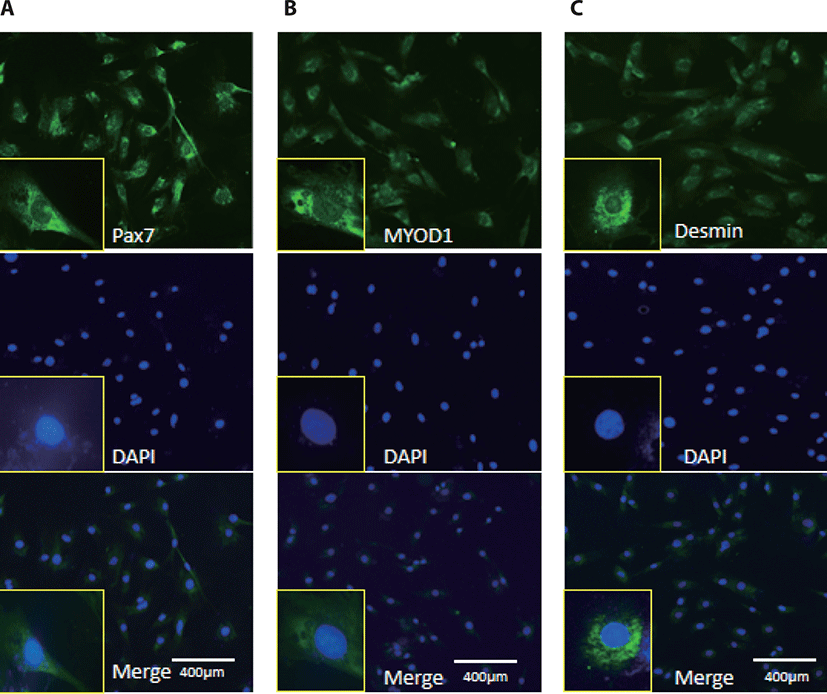
BSC were grown on 14 mm coverslip-bottomed dishes and fixed with 4% paraformaldehyde at ambient temperature for 20 min, washed with phosphate buffered saline (PBS), permeabilized with 0.2% Triton X-100 (Sigma-Aldrich) at ambient temperature for 30 min, and washed with PBS again. The polyclonal anti-myogenic differentiation 1 (MYOD1) (BS-2442R, Bioss, Beijing, China), anti-paired box 7 (Pax7) (AB-528428, Abcam, Cambridge, UK), and anti-desmin Po (BS-20702R, Bioss) were diluted in PBS with 5% bovine serum albumin (BSA) at a ratio of 1:100 and incubated at ambient temperature for 2 h. The horseradish peroxidase (HRP)-labeled Goat Anti-Mouse Immunoglobulin G (IgG) (H + L) (A0216, Beyotime, Shanghai, China) or Goat Anti-Rabbit IgG-HRP antibody (BS-3550R, Bioss) was diluted in PBS with 5% BSA at a ratio of 1:200 and incubated for 2 h at ambient temperature in the dark. Finally, the BSC was incubated in the 0.1 μg/mL 4’,6-diamidino-2-phenylindole, dihydrochloride solution for 30 min in the dark and observed with a fluorescence microscope (WF10X, Olympus, Tokyo, Japan).
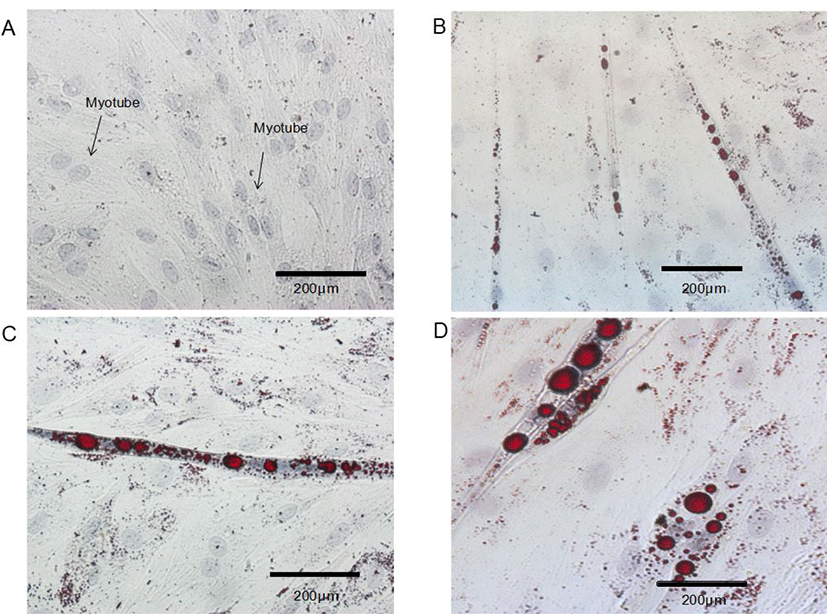
To evaluate the transdifferentiation BSCs, after 96 h of differentiation, the cells were fixed with Oil Red O (ORO) Fixative for 30 min and stained with hematoxylin staining solution for 2 min at ambient temperature. The staining protocol was performed according to the instructions provided by the ORO staining kit (G1262, Solarbio, Beijing, China). Images were collected using an inverted microscope (IX-73, Olympus) and 20 different stained areas were captured. Image analysis followed the protocol of Deutsch et al. [6] using the ImageJ software 1.43u (http://rsbweb.nih.gov/ij). The average lipid droplet size was evaluated using GraphPad Prism 6.07.
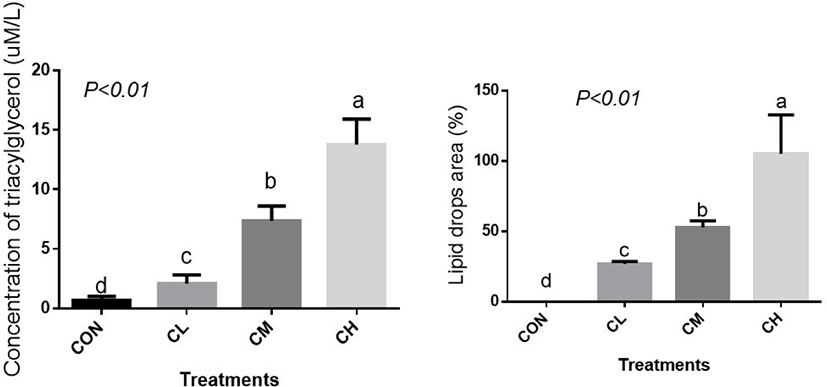
The concentration of triacylglycerol (TAG) was measured using the TAG Quantitative Assay Kit (Applygen, Beijing, China) according to the manufacturer’s instructions. After 96 h of differentiation, the BSC was washed with PBS to remove the medium, and the cells were lysed with Radio-ImmunoPrecipitation Assay lysis solution at ambient temperature for 10 min. Lipase is used to decompose TAG in cell lipid droplets to release glycerol, and the glycerol release amount is measured by spectrophotometric detection at 570 nm.
After BSC was induced to differentiate in vitro for 96 h, total RNA was extracted from cells using Trizol reagent (Thermo Fisher Scientific). The integrity (quantity and quality) of the extracted total RNA was quantified using a NanoDrop ND-100 spectrophotometer (2000C, Thermo Fisher Scientific), and the purity values (A260 / A280) of all RNA samples were greater than 1.80 and were further verified by 1% agarose gel. The RNA samples of good quality were selected for further reverse transcription. Total RNA of 1 μg was reverse-transcribed into cDNA using the FastKing one-step kit (Tiangen Biotech, Beijing, China). Real-time polymerase chain reaction (RT-PCR) was performed using the SYBR premix SuperReal PreMix Plus Kit (SYBRGreen, Tiangen Biotech) and thermocycler Agilent Mx3000/5p system (Agilent Mx3000/5p, Agilent Technologies, Santa Clara, CA, USA). The glyceraldehyde-3-phosphate dehydrogenase gene was used as the housekeeping gene. The genes Pax3, Pax7, MyoD1, and MyoG were selected as myogenic specific marker genes to characterize the extent of myogenesis, while the genes PPARγ, C/EBPα, C/EBPß, sterol regulatory element-binding protein (SREBP)1, stearoyl-coenzyme A desaturase (SCD), carnitine palmitoyltransferase (CPT1), adenosine 5’-monophosphate-activated protein kinase (AMPKα), lipoprotein lipase (LPL), G protein-coupled receptor-43 (GPR43), perilipin-2 (PLIN2), and fatty acid binding protein-4 (FABP4) were used to characterize the degree of adipogenic/lipogenic gene expression. The sequences of the primers are given in Table 1. The thermocycling conditions were as follows: preheating at 95°C for 15 min, a total of 45 cycles of denaturation at 95°C for 10 s, annealing temperature at 60°C for 30 s and extension temperature at 72°C for 32 s with a melting curve from 65°C to 95°C. The quantitative PCR results were calculated using the 2−ΔΔCt method to determine the relative mRNA expression levels.
Pax3, paired box 3; Pax7, paired box 7; MYOD, myogenic differentiation-1; MYOG, myogenin; MYF4, myogenin-4; MRF5, myogenic regulatory factor-5; PPARγ, peroxisome proliferator-activated receptor gamma; C/EBPß, CCAAT/enhancer-binding protein beta; SREBP1, sterol regulatory element-binding protein-1; C/EBPα, CCAAT/enhancer-binding protein alpha; ACC, acetyl-coenzyme A carboxylase; FAS, fatty acid synthetase; CPT1, carnitine palmitoyltransferase; SCD, stearoyl-coenzyme A desaturase; AMPKα, adenosine 5’-monophosphate-activated protein kinase; LPL, lipoprotein lipase; GPR43, G protein-coupled receptor-43; FABP4, fatty acid binding protein-4; PLIN2, perilipin-2; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
After BSC differentiation culture for 96 h, the content of adiponectin in the differentiation medium was analyzed using the bovine adiponectin ADP kit (Mlbio, Shanghai, China). The absorbance (OD value) was measured with a microplate reader (BIO-RAD, Hercules, CA, USA) at a wavelength of 450 nm, and the concentration of bovine adiponectin in the sample was calculated from the standard curve.
After 96 h of BSC differentiation, total RNA was extracted from each treatment using TRIzol™ Reagent (Invitrogen, Waltham, MA, USA) according to the manufacturer’s instructions. The isolated RNA was cleared of contaminating genomic DNA by DNase treatment (Thermo Fisher Scientific). Prior to Illumina RNA sequencing, 1% nondenaturing agarose gel electrophoresis and a Nano-Photometer® spectrophotometer (IMPLEN, Munich, Germany) were used to assess the quality and quantity of the isolated RNA, respectively. Samples satisfying the criteria of RNA integrity number > 7 were utilized for the following library preparation. RNA purification, reverse transcription, library construction, and sequencing were completed by AMOGENE. Briefly, the mRNA was enriched by oligo (dT) beads and was fragmented into short fragments through fragmentation buffer at 94°C for 5 min. First-strand cDNA was synthesized by a random hexamer primer (Illumina) and M-MuLV Reverse Transcriptase (Invitrogen), using the short fragments as templates, at the condition of 25°C for 10 min, followed by 42°C for 50 min for synthesis and 70°C for 15 min for RTase inactivation. Second strand cDNA synthesis was subsequently performed using Second Strand Master Mix (Illumina) at 16°C for 1 h. Next, the cDNA fragments of 150–200 bp in length were treated with T4 DNA polymerase and 5′ phosphorylated, and then an adenine (A) residue was added to the 3′ ends of the DNA. Moreover, adapters were also ligated to the ends of these target template DNAs. After ligation, the cDNA library was obtained by PCR amplification (5–9 cycles). Finally, PCR products were purified, and library quality was evaluated on an Agilent. Bioanalyzer 2100 system (Agilent Technologies). The library preparations were sequenced on an Illumina Hiseq 4000 platform using paired-end technology following the manufacturer’s standard guidelines. Fastp was used to filter the original sequencing data to obtain clean data, and the filtered data were evaluated with the RSeQC software package to obtain high-quality sequencing data, and then Spliced Transcripts Alignments to a Reference software was used for sequence alignment analysis with the reference genome.
Cuffdiff was used to assemble the RNA-Seq alignments and estimate gene expression and transcript. The edgeR package was used for the identification of differentially expressed genes (DEGs) within samples. Genes with statistical significance were retained first (q-value < 0.05); significant DEGs were compared with a fold change of > 1.5 than 1.5 or < 0.667 were retained [log2 (1.5) = 0.5849; log2 (0.667) = −0.5849].
The DEGs were subjected to the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway and enrichment analysis of Gene Ontology (GO). KEGG enrichment analysis was to identify the pathways of significant enrichment of DEGs, gene function, genomic information, and functional information. The GO classification includes the following three aspects: biological process (BP), molecular function (MF), and cellular component (CC), which define and describe the functions of selected genes and corresponding proteins. KEGG was separately generated by (http://www.genome.jp/kegg).
One-way ANOVA with multiple comparisons test was used to evaluate the statistical significance of differences among treatments. All data are presented as mean ± SEM. The graphs were prepared using GraphPad Prism 6.07 (GraphPad, San Diego, CA, USA). Statistical significance was set at p < 0.05.
RESULTS
MyoD1 antibodies, Desmin, and Pax7 antibodies were used to monitor myogenic differentiation of BSC, which has been characterized by immunofluorescence staining of proliferation positive BSC (Fig. 1).
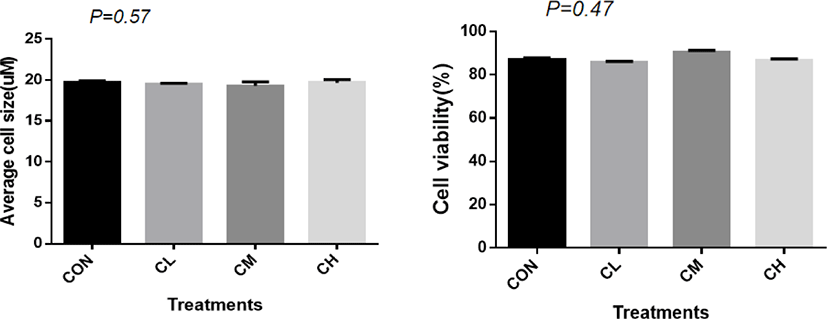
A total of 1,125 ± 7.82 cells were analyzed for each treatment to measure the average cell size. There was no significant difference in cell size (p = 0.57) and cell vitality between treatments (p = 0.47) (Fig. 2).
Control BSC proliferated and fuse to form myotubes. Upon incubation of BSC with ciglitazone, the cells became adipocyte-like cells, and the effect is obvious after 96 h (Fig. 3). Increasing the concentration of ciglitazone greatly accelerated the appearance of adipocyte-like cells, and strongly increased the number of BSC that differentiated into adipocytes. Exposure of post-confluent cells to the differentiation medium plus ciglitazone decreased myotube formation, with a concomitant increase in the area of cells containing lipid droplets and the formation of adipocytes.
The concentration of cellular TAG, the main component of lipid droplets, was elevated linearly by CL, CM, and CH treatments and, correspondingly, CL, CM, and CH treatments caused a profound increase in the percent area of lipid droplets (Fig. 4) (p < 0.01).
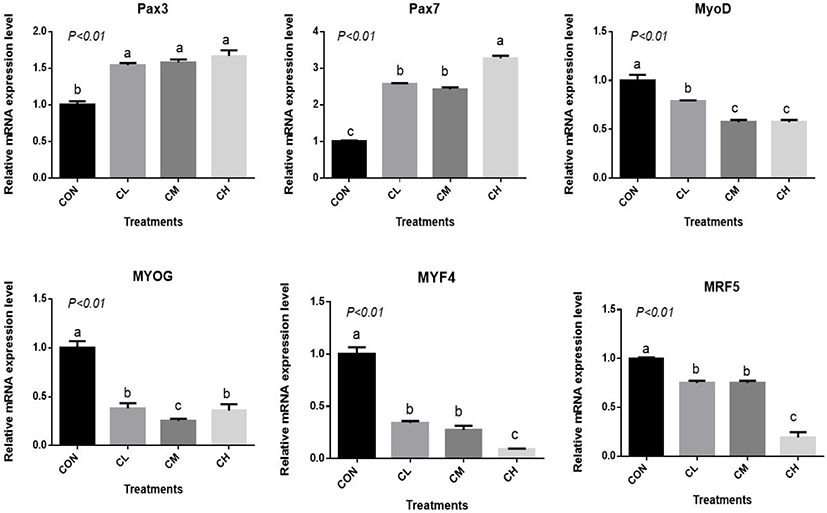
The CL, CM, and CH treatments increased Pax3 and Pax7 expression and decreased MYOD, MYOG, myogenin-4 (MYF4), and myogenic regulatory factor-5 (MRF5) expression relative to CON (p < 0.01) (Fig. 5). The expression of MYF4 and MRF5 were the least in the CH-treated BSC (p < 0.01).
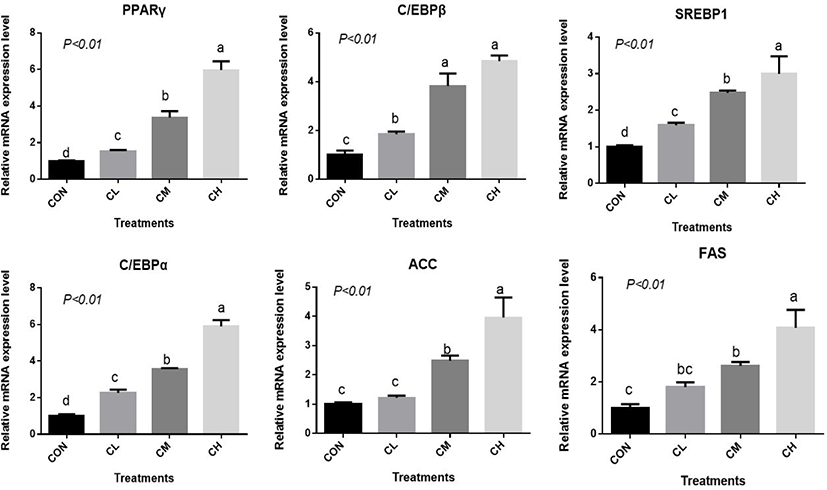
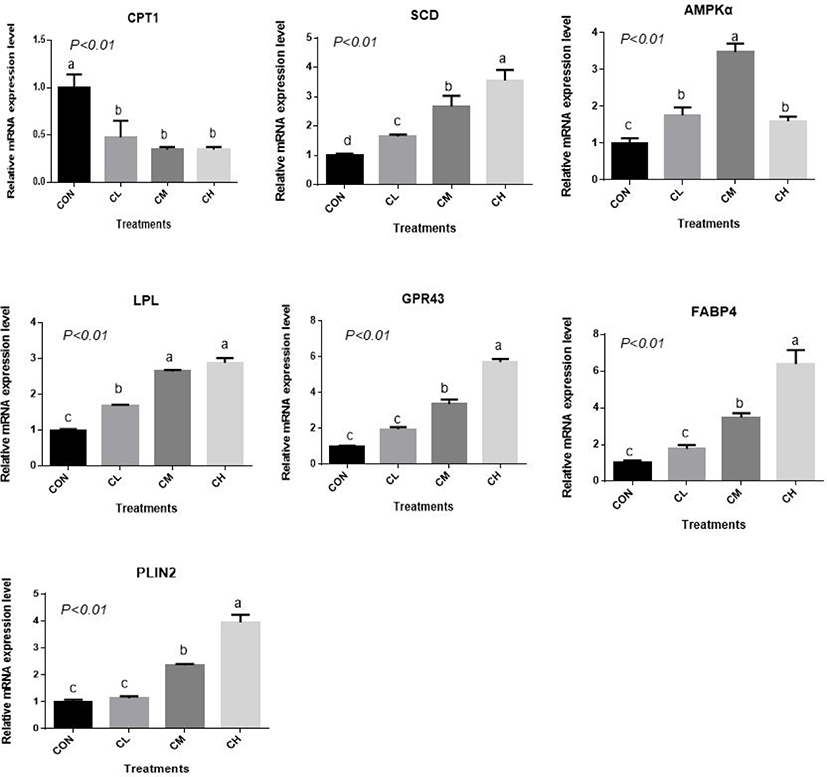
There was a dose-response to ciglitazone addition for the expression of adipogenic transcription factors PPARγ, SREBP1, C/EBPα, C/EBPβ, acetyl-coenzyme A carboxylase, and fatty acid synthetase (p < 0.01), and the greatest increase in the expression of these genes, were caused by CH-treated BSC (Fig. 6). Relative expression of SCD, LPL, GPR43, FABP4, and PLIN2 was dose-dependent as the addition level of ciglitazone increased (p < 0.01) (Fig. 7). Expression of CPT1 was depressed by the CL, CM, and CH-treated BSC stepwise relative to CON BSC. Expression of AMPKα (p < 0.01) was greater in CL, CM, and CH-treated BSC than in CON BSC, and the CM treatment caused the greatest increase.
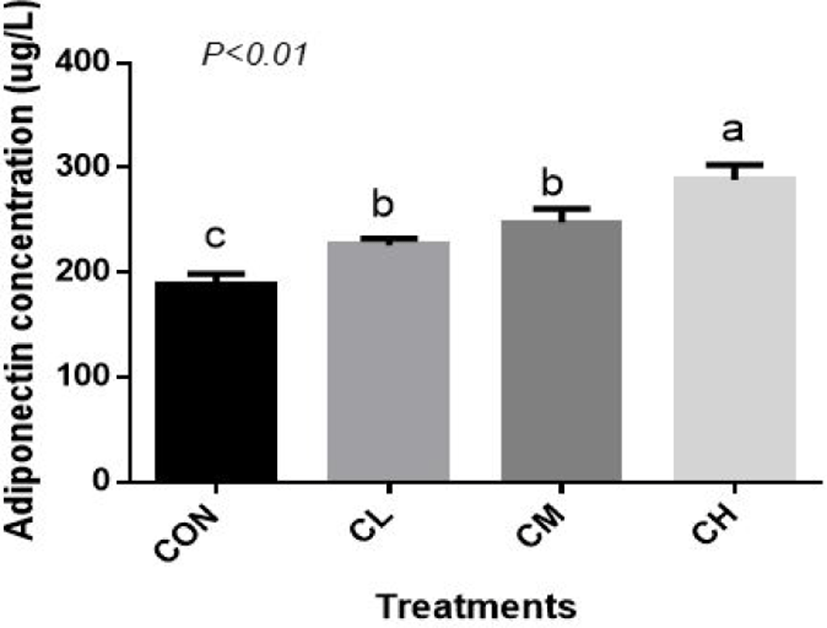
Compared with CON BSC, the CL, CM, and CH treatments increased the amount of adiponectin (ADP) secretion (p < 0.01), and the highest concentration of ADP was caused by the CH treatment (Fig. 8).
Whole-genome expression profiling with RNA-Sequencing analysis revealed significant DEGs when the BSC were treated with different levels of ciglitazone. Compared with CON, the CL treatment up-regulated 611 genes and down-regulated 510 genes; the CM treatment up-regulated 565 genes and down-regulated 527 genes; whereas the CH treatment up-regulated 501 genes and down-regulated 435 genes (Table 2). Combined with the analysis of these results of differential expressed genes, BSC treated with CL, CM, and CH had 281 DEGs as shown in the volcano map (Fig. 9A, B, and C).
| Type | Treatment | ||
|---|---|---|---|
| CL | CM | CH | |
| Up | 609 | 562 | 501 |
| Down | 508 | 526 | 432 |
| Total | 1,117 | 1,088 | 933 |
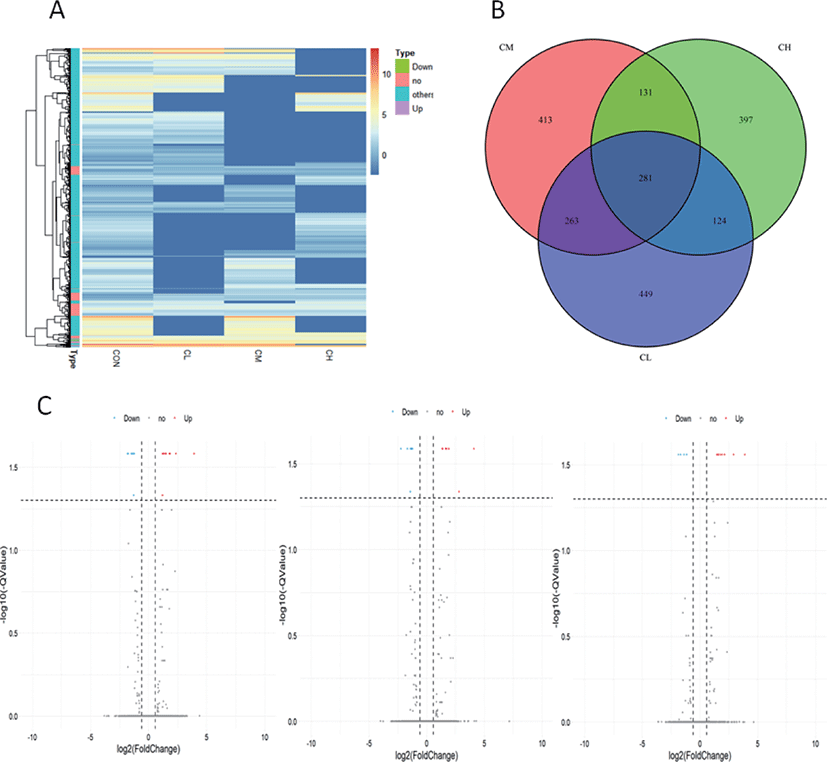
Pathway analysis of DEGs was performed to understand the biological relationship between pathways and molecules. The GO annotation was classified into three clusters: BP, MF, and CC. The 10 top significantly enriched GO terms in which the calculated by the hypergeometric test was Q < 0.05 were selected. As shown in Fig. 10 and (Supplementary Table S1, biological pathways were significantly changed by GO terms of ciglitazone-treated BSC. Top 10 GO enrichment analyses significantly changed cell proliferation, protein trimerization, fat cell differentiation, cellular response to external stimulus, positive regulation of release of cytochrome c from mitochondria, and type B pancreatic cell proliferation process. Molecular functions such as nuclease activity, endonuclease activity, deoxyribonuclease activity, growth factor activity, kinase inhibitor activity, biotinidase activity, and receptor-ligand activity were up-regulated by ciglitazone.
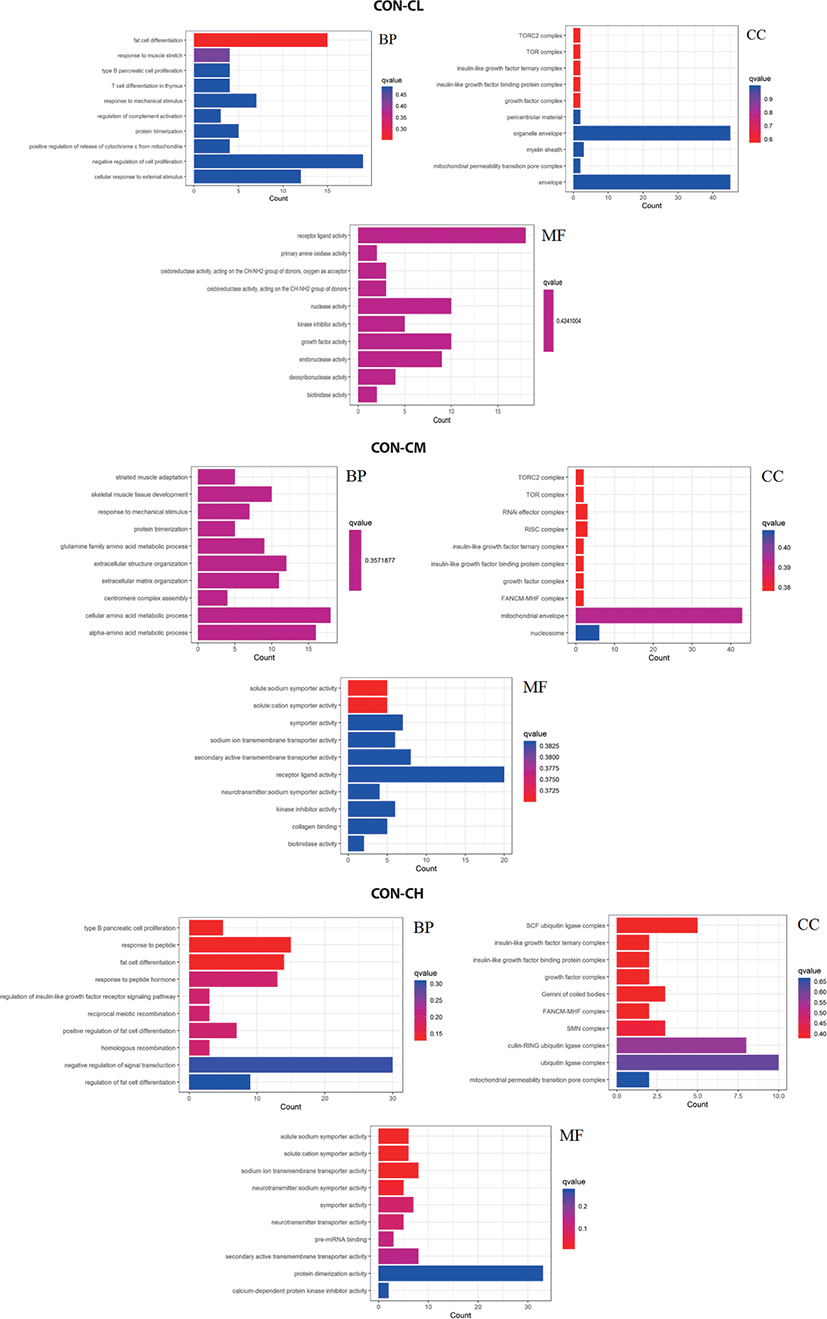
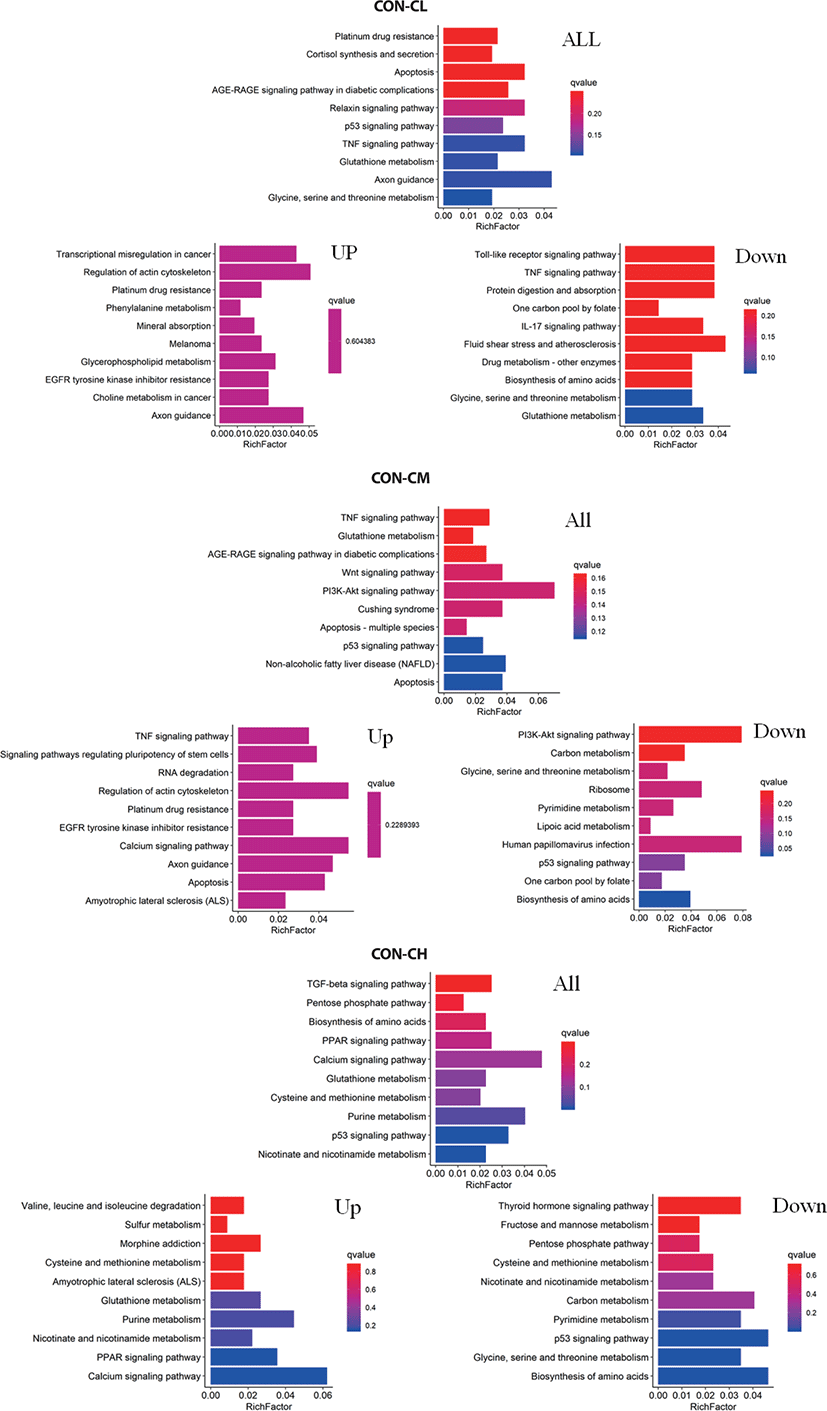
Pathway analysis of DEGs was conducted to understand the pathways and molecular interactions involved with ciglitazone treatment. The 10 top significantly enriched GO terms in which the calculated by the hypergeometric test was Q < 0.05 were selected. Enrichment degree was identified by Gene Ratio, P. adjust, and enriched genes. In the CL treated BSC, KEGG enrichment analysis indicated that a total of 465 DGEs were enriched in 31 pathways and mainly in glycine, serine, and threonine metabolism, tumor necrosis factor (TNF) signaling pathway, and the p53 signaling pathway. Genes with up-regulated expression were mainly enriched in mineral absorption, transcriptional misregulation in cancer, choline metabolism in cancer, glutathione metabolism. Genes with down-regulated expression were mainly enriched in the TNF signaling pathway, biosynthesis of amino acids, protein digestion, and absorption.
In the CM treated BSC, a total of 484 DGEs were enriched in 18 pathways and mainly in apoptosis, p53 signaling pathway, non-alcoholic fatty liver disease (NAFLD), PI3K-Akt signaling pathway, Wnt signaling pathway, and TNF signaling pathway. Genes with up-regulated expression were mainly enriched in TNF signaling pathway, the calcium signaling pathway, apoptosis, RNA degradation, and genes with down-regulated expression were mainly enriched in the biosynthesis of amino acids, p53 signaling pathway, ribosome, carbon metabolism, and the PI3K-Akt signaling pathway. In the CH treated BSC, a total of 397 DGEs were enriched in 30 pathways and mainly in the p53 signaling pathway, PPAR signaling pathway, biosynthesis of amino acids, pentose phosphate pathway, and TGF-beta signaling pathway. Genes with up-regulated expression were mainly enriched in the calcium signaling pathway, PPAR signaling pathway, purine metabolism, nicotinate and nicotinamide metabolism, and genes with down-regulated expression were mainly enriched in the biosynthesis of amino acids p53 signaling pathway, glycine, serine, and threonine metabolism, pyrimidine metabolism, and carbon metabolism (Fig. 11 and Supplementary Table S2).
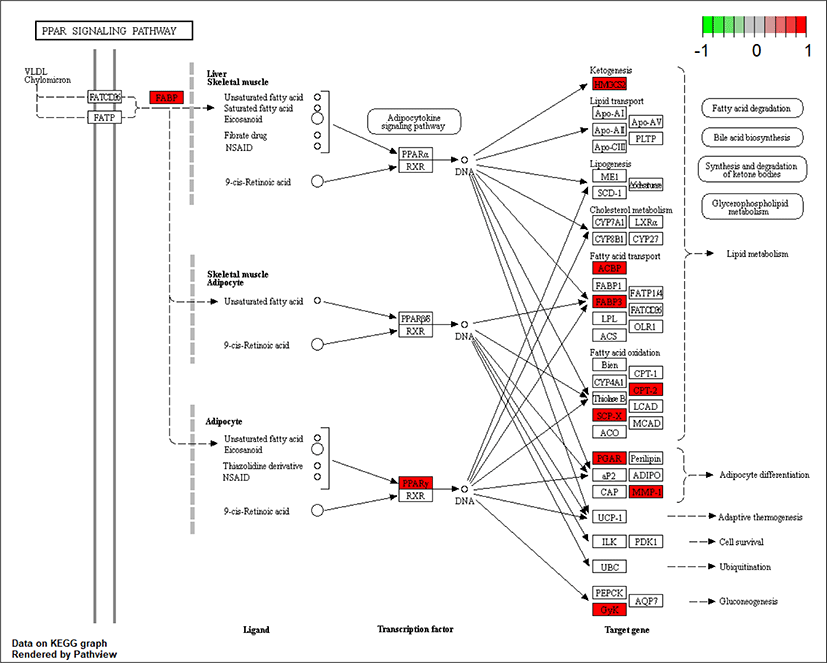
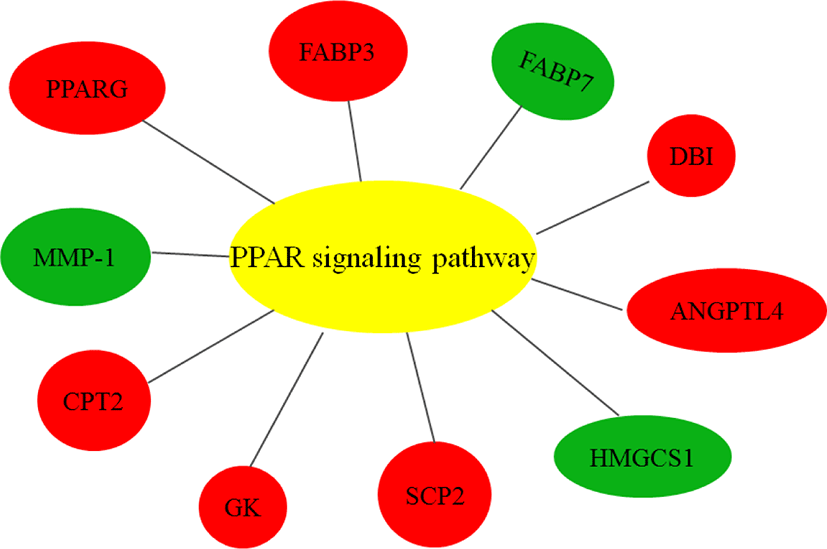
As ciglitazone is a PPARγ agonist, we analyzed the PPAR-signaling pathway enriched by DEGs during adipogenic trans-differentiation of BSC induced by different concentrations of ciglitazone (Fig. 12). The high-concentration of ciglitazone significantly up-regulated the PPAR signaling pathway, there were 10 genes differentially enriched in this pathway, and the proportion of differentially enriched genes was 10/397. Pathway analysis of differentially enriched genes indicated 7 up-regulated genes and 3 down-regulated genes. Based on the up-regulated expression of PPARγ, the downstream fatty acid transport-related genes (diazepam binding inhibitor [DBI], FABP3), fatty acid oxidation-related genes (CPT-2, sterol carrier protein 2, FABP3), adipocyte differentiation-related genes (angiopoietin like 4, glycogen neogenesis gene glucokinase), and the adipocyte differentiation-related gene FABP7 were up-regulated. The expression of matrix metalloproteinase-1 and the ketogenic gene hydroxymethylglutaryl-CoA synthase2 was down-regulated (Fig. 13).
DISCUSSION
This study addressed the hypothesis that ciglitazone as a PPARγ ligand in the absence of an adipocyte differentiation cocktail would increase adiponectin and adipogenic gene expression in BSC. Previous studies investigated TZD as an extrinsic factor that influences myogenic satellite cells by inducing the expression of several transcription factors, including PPARγ and C/EBPα [7,8]. In cases where the conditions of the adipocytes containing the PPARγ activator permit, these cells can differentiate into mature adipocytes [9]. Li et al. [10] assumed that myoblasts and adipocytes have similar bio-initiating sources, and the potential for trans-differentiation to produce marbling or intramuscular lipogenesis was hypothesized. Transcriptional control of adipocyte differentiation requires a series of continuous gene expression events and activation of many key signaling pathways [11]. Cell lipid accumulation and morphological changes accompanying lipogenesis are caused by the induced expression of specific genes during differentiation [12]. In the present study, the decrease in the myotube formation and the increase in the accumulation of lipid droplets caused by ciglitazone addition in BSC. This may indicate that ciglitazone could induce the BSC to differentiate into adipocytes.
Studies have reported that many transcription factors regulate adipocyte differentiation, such as C/EBPs and PPARs [9]. Muscle satellite cells can be trans-differentiated into mature adipocytes by ectopic expression of adipose trans-differentiation factors such as PPARγ [13]. Previous studies had documented that when the PPARγ gene was overexpressed in G8 myoblasts under optimal culture conditions for muscle differentiation, the expression levels of myogenic factors MRF5, MYOD, and MYF4 were depressed, and the myoblasts could not differentiate into myotubes [14]. In mammalian cells, PPARγ and C/EBPα are major regulators of lipogenesis and be transcription targets [15]. PPARγ is induced during the differentiation of preadipocytes into adipocytes, which is critical for precursor cells to differentiate into mature adipocytes [16]. Adipocyte differentiation also involves the expression of several other transcription factors that interact at different the process of adipogenesis to produce mature adipocytes [17]. Teboul et al. [4] demonstrated that TZDs specifically bind to and activate PPARγ, increase the expression of PPARγ and related adipokines, and promote adipocyte differentiation. Kageyama et al. [18] demonstrated that pioglitazone increased body weight by selectively stimulating PPARγ and promoting lipid storage in white adipose tissue. Lizcano [19] reported that JMJD2C could inhibit PPARγ activity and reduce triglyceride accumulation in adipocytes. Although PPARγ and C/EBPα are highly synergistic in the adipogenic process, adipocyte genes vary in their reliance on C/EBPα and PPARγ [20].
Pax3 and Pax7 are markers of the differentiation of mesenchymal cells toward a myogenic cell lineage. Up-regulation of the expression of Pax3 and Pax7 increases the expression of myogenic regulatory factors [21], which include MYOD and MYOG. Up-regulation of MYOD expression promotes the conversion of somite-derived cells into myoblasts; MYOG is involved in the fusion of myoblasts to form myotubes [22]. Cornelison et al. [23] identified MYOD as a key negative regulator of brown adipocyte development. Similarly, Wang et al. [24] reported that MYOD negatively regulated the f brown adipocyte development and concluded that loss of MYOD facilitates adipogenic trans-differentiation of myoblasts in vivo. The PLIN2 is primarily expressed during the early stages of adipocyte differentiation, which promotes lipid droplet accumulation in muscle cells [25]. Studies with 3T3-L1 adipocytes indicated that PLIN2 could be used as a marker gene for precursors during adipocyte differentiation [26]. In this study, ciglitazone in the absence of an adipocyte differentiation cocktail also induced an up-regulation of adipogenic gene expression accompanied by low expression of MYOD, and the higher PLIN2 expression and adiponectin promotes the higher lipid droplets accumulation in the BSC [3].
Whole-genome RNA-Seq GO analysis and KEGG enrichment analysis of DEGs showed that most DEGs were closely related to cell proliferation, cell differentiation, and cell metabolic processes. PPAR signaling pathway, P53 signaling pathway, Wnt signaling pathway, ribosome, signaling pathway regulating pluripotency of stem cells, TNF signaling pathway, and calcium signaling pathway participate in the adipogenic transdifferentiation process of BSC. The high concentration of ciglitazone significantly up-regulated the PPAR signaling pathway, and 7 out of 10 genes were up-regulated in this pathway. Besides, the increase in the accumulation of lipid droplets and the concentration of TAG and adiponectin further indicated the regulatory role of PPARγ in adipogenic transdifferentiation of BSC.
The PI3K-Akt signaling pathway plays an important regulatory role in adipocyte differentiation and adipogenesis. Inhibiting the activation of PI3K-Akt could depress adipogenesis and differentiation, and down-regulate the expression of adipogenic genes PPARγ, C/EBPa, FABP4, and FATP1 [27,28]. Wnt signaling has been reported to inhibit adipogenesis. Adipose tissue contains adult stem cells (ADSCs) similar to mesenchymal stem cells, which inhibit adipogenic differentiation by activating the Wnt signaling pathway [29–31]. In this study, the RNA sequencing results revealed that the low concentration of ciglitazone could down-regulated the genes in the regulation of the Wnt signaling pathway, which revolved in adipogenesis and lipid metabolism.
TGF-β1 is a factor activated by inflammation and participates in the process of chronic fibrosis of organs. TGF-β1 transmits fibrotic signals through its downstream kinase substrate Smads protein. The TGF-β1/Smad3 signaling pathway and organ fibrosis are closely related. TGF-β1 binds to its receptor on the cell membrane causes intracellular Smad2 and Smad3 phosphorylation and Smad4 binds to form a complex that enters the nucleus and cooperates with other transcription factors to regulate the expression of genes related to cell proliferation and differentiation [32–35]. In our study, the high concentration of ciglitazone can inhibit the activation of the TGF-β1/Smad3 signaling pathway. This may suggest that ciglitazone could promote the adipocyte phenotype in BSC through these signaling pathways. These results indicate that ciglitazone as a PPARγ agonist can promote lipid droplet formation and triglyceride accumulation in BSC, and effectively increase adiponectin concentration and adipogenic gene expression. RNS-seq showed that treatment with different concentrations of ciglitazone significantly affected the p53 signaling pathway, PPAR signaling pathway, TNF signaling pathway, PI3K-Akt signaling pathway, and Wnt signaling pathway.
In conclusion, our results support at least partial conversion of BSC to adipocytes by ciglitazone in the absence of an adipocyte differentiation cocktail. Further experiments are needed to elucidate the molecular mechanisms by which PPARγ affects lipogenesis during the process of adipogenic differentiation of BSC and the regulatory pathways that influence other regulators of lipid production.