INTRODUCTION
Comprehensive efforts have been directed toward elucidating the ecological and metabolic features of methanogenic archaea to inform effective strategies for mitigating methane emissions in livestock production systems. Methanogenic archaea inhabit a range of anoxic environments including the rumen, marine sediments, wetlands, and deep-sea hydrothermal vents, where their dominant methanogenic pathways, primarily hydrogenotrophic and methylotrophic, vary according to ecological context [1,2]. However, many methanogens remain difficult to isolate and cultivate under laboratory conditions, resulting in a knowledge base that is skewed toward in vitro data from non-rumen habitats and remains fragmentary with respect to rumen-specific biology [3]. Within the rumen, methane production is strongly influenced by methanogen diversity, hydrogen flux, and syntrophic interactions with bacterial guilds, yet the phylogenetic and functional relationships remain poorly defined [4,5].
Comparative pangenome analysis provides a genome-wide framework for dissecting these relationships, enabling gene-level resolution of phylogenetic, functional, and metabolic divergence across diverse taxa. Notably, the Zoonomia Consortium’s alignment of 240 mammalian genomes has established a methodological benchmark for large-scale comparative analysis, integrating large-scale genome alignment, normalization, and statistical inference in a unified analytical pipeline [6]. Building upon such advances in large-scale genome comparative analysis, similar efforts in the field of animal microbiology have followed suit. The Hungate1000 project provided foundational resources by sequencing 410 cultured rumen microbial genomes, which were later expanded by the assembly of 913 metagenome-assembled genomes (MAGs) from bovine rumen metagenomes and the construction of a 4,941-member Rumen Uncultured Genome catalogue, expanding the landscape of species- and function-level variation and strengthening quantitative links to methanogenesis pathways [7,8]. More recently, genome-resolved surveys have expanded archaeal resources and environmental breadth, including a catalogue of 998 unique ruminant-gut archaeal genomes across 10 host species and large MAG datasets from Nelore cattle, which together sharpen genome-level resolution of rumen methanogenesis [9,10]. Despite these advances, several critical questions remain unanswered. It still lacks a genome-resolved understanding of how methanogen gene repertoires vary across habitats and how these differences map onto phylogenetic lineages, functional capacities, and methanogenic strategies within the rumen. Furthermore, there is no integrated framework that connects methanogen genomic features with co-occurring microbial guilds that either supply reductants or act as metabolic competitors within the methanogenesis network.
To address these knowledge gaps, we employed a two-stage comparative genomics approach integrating phylogenetic reconstruction, pangenome analysis, and metagenomic profiling to systematically characterize rumen methanogens and their ecological interactions. Initially, we constructed phylogenetic trees using 16S rRNA and the key methanogenesis marker gene mcrA from methanogens isolated across diverse ecosystems, and performed habitat-stratified pangenomic comparisons to identify gene-level signatures and clustering patterns of each habitat. And then, we investigated rumen shotgun metagenomic data to delineate candidate methane substrate producers, consumers, and competitor lineages within the methanogenic network, thereby nominating functionally relevant targets for methane mitigation in rumen ecosystems [11].
MATERIALS AND METHODS
Genomic sequences were retrieved primarily from GenBank, with curated versions cross-validated via RefSeq. Accession numbers, version identifiers, BioProject, BioSample, TaxID, and download dates are detailed in Supplementary Table S1. The final dataset included 71 methanogen reference genomes, spanning 10 from the rumen, 4 from the human gut, 17 freshwater, 14 sewage, 20 seawater, 4 soil, and 2 cockroach-derived strains. For 16S rRNA-based phylogeny, rRNA loci were predicted and extracted using Barrnap v0.9, followed by coordinate-based trimming to remove incomplete ends. Multiple sequence alignment was performed using MAFFT v7 [12]. For functional phylogeny, the mcrA gene (encoding methyl-coenzyme M reductase subunit A) was detected via HMMER v3 searches against the corresponding Pfam domain, retaining only full-length hits in line with established use of mcrA as a methanogen marker. The resulting amino-acid sequences were aligned with MAFFT v7 [13–15]. Maximum-likelihood phylogenies were inferred using IQ-TREE v2, and annotated phylogenetic trees were visualized with iTOL v6 [16,17].
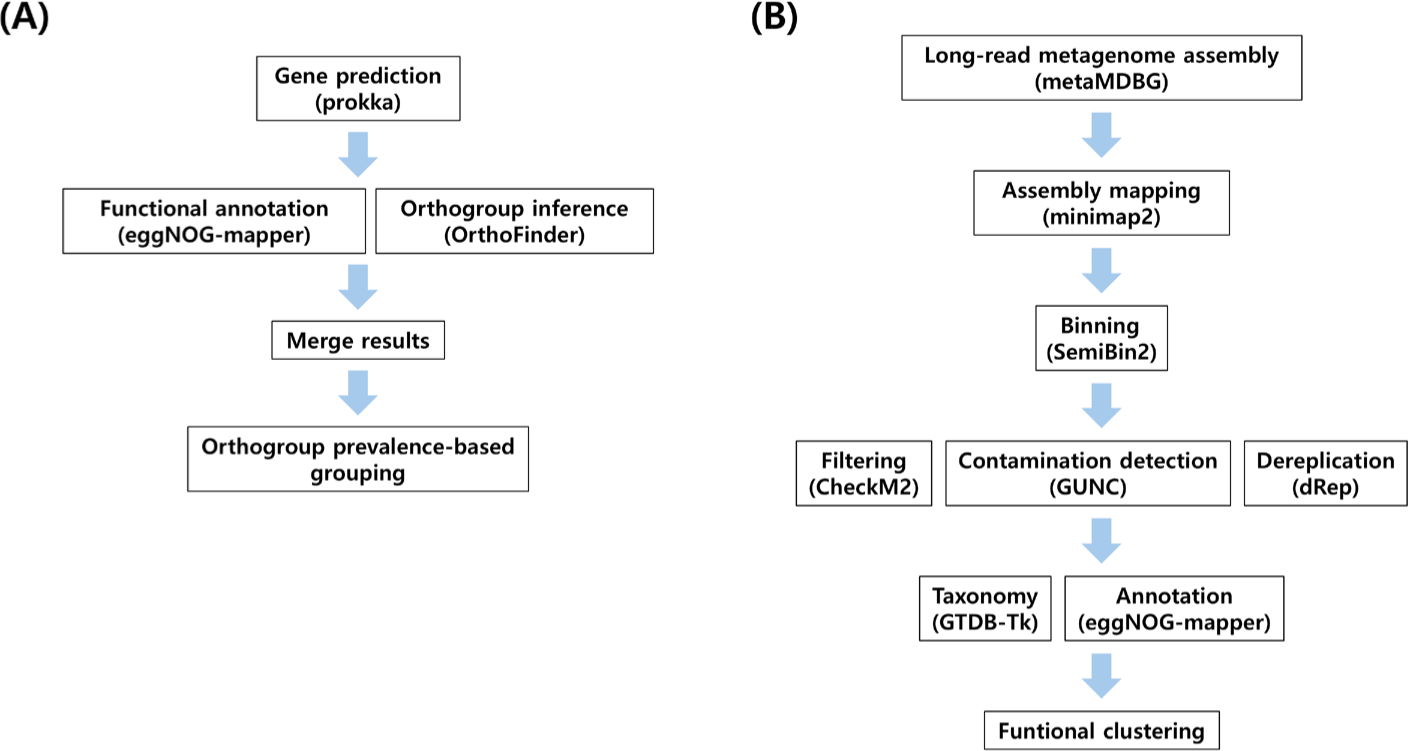
Pangenome analysis from diverse ecosystems was conducted following established protocols with minor modifications [11]. The analytical workflow is summarized in Fig. 1A. Protein-coding genes were predicted and functionally annotated from assembled genomes with Prokka v1.14.6 [18]. The resulting translated proteomes were used for orthogroup inference with OrthoFinder v3.1.0, and orthogroups (COG) presence–absence matrices were used to delineate core, shell and cloud gene sets following standard pangenome definitions [19]. Functional annotation of protein sequences was assigned using eggNOG-mapper v2.1.13 against the eggNOG v5 orthology database. KEGG orthology (KO) and gene ontology (GO) assignments derived through the eggNOG framework were used to functionally characterize orthogroups [20,21]. The combined outputs from OrthoFinder and eggNOG-mapper provided gene clusters with consolidated functions, which formed the basis for downstream interpretation of methanogen pangenome structure across environments [19]. Data visualization was carried out using Seaborn v0.13.2 and Matplotlib v3.10.6 [22,23]. The COG were scored as present or absent per strain and classified by prevalence as follows: core, ≥ 99% of genomes; soft-core, ≥ 95% and < 99%; shell, ≥ 15% and < 95%; cloud, < 15% [24]. Results for the cloud set were omitted.
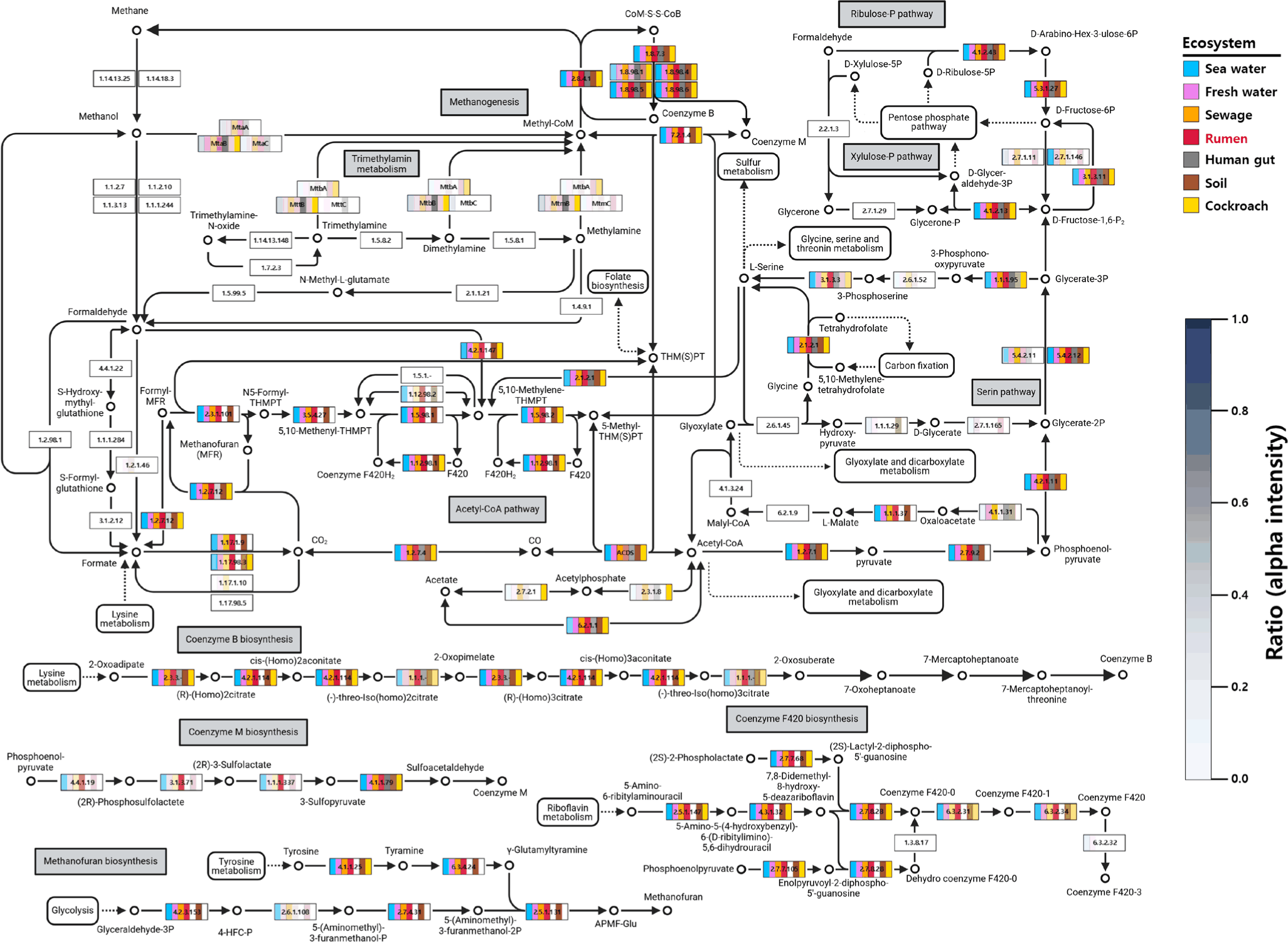
From eggNOG-mapper v2.1.13 annotations of the methanogen reference genomes used in the pangenome analysis, KEGG Orthology assignments linked to the methanogenesis pathway ko00680 were retrieved and compiled. A KO was deemed present if at least one encoded protein was annotated with the respective identifier. Environment-specific KO frequency was calculated as the proportion of genomes in each habitat containing the KO. The resulting KO-by-environment matrix was visualized both as pathway tile maps and matrix plots using Matplotlib v3.10.6.
Schematic workflows of rumen MAGs functional analysis were illustrated in Fig. 1B. Shotgun metagenome reads were obtained from dairy cow rumen fluid sample sourced from previously published datasets by Kang et al [25]. Long-read metagenome assembly was performed with metaMDBG v1.2 [26]. Read mapping and coverage profiling were performed using minimap2 v2.30. Binning was conducted with SemiBin2 v2.2.0 [27]. Genome quality was evaluated with CheckM2 v1.1.0 and contamination was filtered using GUNC v1.0.6. Bins with ≥ 50% completeness and < 10% contamination were retained [28–30]. Nonredundant representatives were obtained by dereplication with dRep v3.6.2 [31]. Taxonomic assignment used GTDB-Tk v2.4.1 with GTDB reference data release r226 [32]. Functional annotation of predicted protein sequences was performed using eggNOG-mapper v2.1.13. MAGs were categorized into four ecological roles using curated marker genes.
Methanogens were identified as archaeal MAGs encoding the methyl-coenzyme M reductase operon (mcrA/B/G), optionally supported by mtrA/B/E, which are canonical to methanogenic energy metabolism [33,34]. Substrate supply producers were defined as MAGs encoding one or more routes that provide key methanogenic substrates: H2 production via [FeFe]-hydrogenase (hydA) together with its maturation genes (hydE/F/G); formate production via pyruvate-formate lyase (pflA/pflB); acetate production via the phosphotransacetylase–acetate kinase pair (pta/ackA) [35,36]. Competitive sinks-competitors captured respiratory pathways that divert the same reductants (H2/electrons) away from methanogenesis. Dissimilatory nitrate reduction to ammonium (DNRA) was identified by nrfA/H. The reductive Wood–Ljungdahl pathway (WLP) evidenced by cooS (CODH), with fhs and metF treated as supportive folate-branch markers rather than strict requirements [37–39]. Methanotrophs were identified by the presence of the particulate methane monooxygenase gene pmoA, a widely used functional and phylogenetic marker [40,41]. Out of 903 initial bins from SemiBin2, 151 passed CheckM2 filtering, 116 passed GUNC quality control, and 106 dereplicated MAGs remained. Ultimately, 67 MAGs with functional roles were used in downstream analysis.
Differential distribution of orthogroups by clade and habitat was assessed using two-sided Fisher’s exact tests (2 × 2 contingency tables) with Benjamini-Hochberg FDR correction (q ≤ 0.05). Enrichment was interpreted via odds ratios (OR): OR > 1 denoting enrichment, OR < 1 indicating depletion. Clade-specific orthogroups were defined as those present in ≥ 80% of strains within a clade and ≤ 20% outside it [42,43].
RESULTS
Comparative phylogenetic analysis based on 16S rRNA and mcrA sequences identified two clades exhibiting substantial overlap, with Jaccard indices exceeding 0.7. The first clade, comprising strains from freshwater and seawater habitats, showed a Jaccard index of 0.75 and included NC_009051.1_Methanoculleus_marisnigri_JR1, NC_009712.1_Methanoregula_boonei_6A8, NZ_AP019781.1_Methanoculleus_chikugoensis_strain_MG62, NZ_CP109831.1_Methanoculleus_submarinus_strain_DSM_15122, NZ_CP113361.1_Methanogenium_organophilum_strain_DSM_3596, and NZ_JOMF01000012.1_Methanomicrobium_mobile_DSM_1539. A second clade, spanning freshwater and sewage-derived isolates, yielded a Jaccard index of 0.714 and consisted of NC_007796.1_Methanospirillum_hungatei_JF-1, NC_018227.2_Methanoculleus_bourgensis_MS2, NC_019943.1_Methanoregula_formicica_SMSP, NZ_CP036172.1_Methanofollis_aquaemaris_strain_N2F9704, and NZ_CP091092.1_Methanomicrobium_antiquum_strain_DSM_21220.
No additional clades showed concordance above the 0.7 threshold. The lack of clade concordance between the relatively conserved 16S rRNA tree and the mcrA tree indicates accelerated sequence divergence in mcrA. Moreover, the failure of environment-based clustering to persist on the mcrA phylogeny points to frequent horizontal gene transfer events and inherent diversification of this functional marker (Fig. 2).

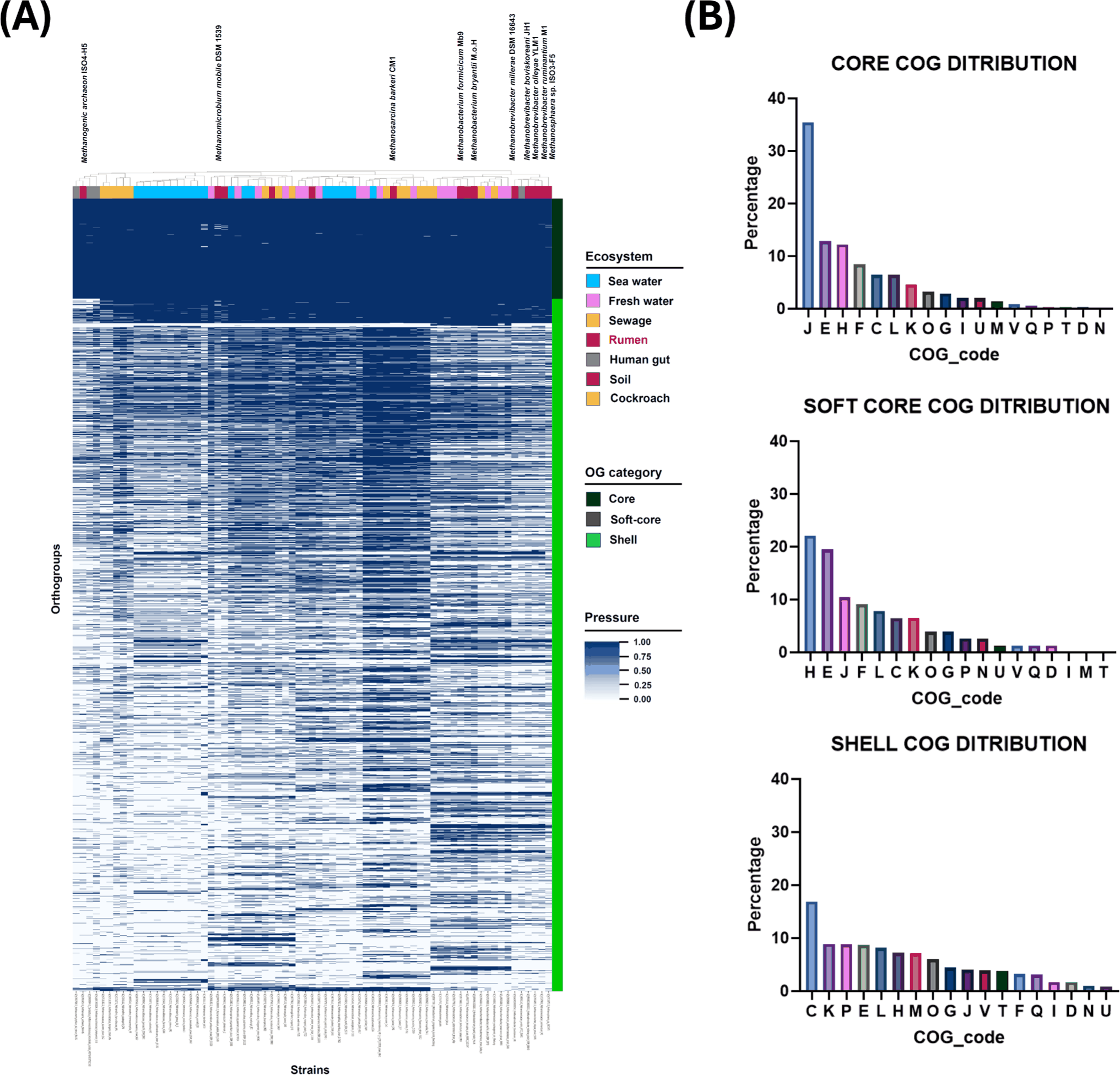
From the 71 methanogen genomes analyzed, a total of 8,695 orthogroups were identified and classified into core (≥ 99% genomes), soft-core (≥ 95% and < 99%), and shell (≥ 15% and <95%) components. This yielded 385 core, 94 soft-core, and 2,573 shell orthogroups. The most abundant COG categories within each group were as follows: J (translation, ribosomal structure and biogenesis; 35.4%), E (amino acid transport and metabolism; 12.8%), and H (coenzyme transport and metabolism; 12.2%) for core; H (22.1%), E (19.5%), and J (10.4%) for soft-core; and C (energy production and conversion; 16.9%), K (transcription; 8.9%), and P (inorganic ion transport and metabolism; 8.8%) for shell (Fig. 3B). These distributions align with prior archaeal pangenomic surveys, emphasizing conserved functions in translational machinery and cofactor metabolism across core genomes [44]. In contrast to typical bacterial patterns, where J, E, and H frequently appear alongside C and F in the core at comparable proportions, the methanogen set here places energy metabolism and nucleotide transport and metabolism predominantly outside the core. The core was enriched for COG J, E, and H, contrasting with the bacterial tendency to balance J, E, and H with C and F at comparable proportions. Therefore, these results support strong conservation of the information-processing machinery in archaea and suggest an elevated fraction of poorly annotated proteins reflecting limited study coverage [44–46]. Additionally, rare or composite COG, including IQ, FG, NU, DJ, DZ, EGP, BQ, and A, were uniquely found within the shell COG. Within the rumen-enriched clade including NZ_CP118753.2_Methanosphaera_sp._ISO3-F5; NC_013790.1_Methanobrevibacter_ruminantium_M1; NZ_CP014265.1_Methanobrevibacter_olleyae_strain_YLM1; NZ_FMXB01000001.1_Methanobrevibacter_millerae_strain_DSM_16643; NC_009515.1_Methanobrevibacter_smithii_ATCC_35061; NZ_BAGX02000054.1_Methanobrevibacter_boviskoreani_JH1, enrichment testing at q ≤ 0.05 showed COG J to be clade-specifically depleted (OR = 0.08), whereas COG M (Cell wall/membrane/envelope biogenesis) was relatively enriched (OR = 2.46; Fig. 3C). The enrichment of cell wall, membrane and envelope functions in the rumen methanogen clade, relative to other clades, supports the interpretation that these taxa engage in potential ecological and physical interactions with coexisting rumen microbiome [47].
KEGG pathway ko00680 was used to assess methanogenesis gene distribution. The methyl-coenzyme M reductase complex, a hallmark of methanogenic metabolism, was universally present across all genomes. Other core methanogenesis enzymes such as heterodisulfide reductase and ferredoxin reductase, also showed complete conservation across environments. However, human gut–derived methanogens exhibited substantial depletion of genes involved in the biosynthesis of coenzyme M, coenzyme B, coenzyme F420, and methanofuran, including K00200, K00201, K00202, K00203, K00205, K00319, K00320, K00441, K00577, K00578, K00579, K00580, K00581, K00584, K00672, K01499, K06914, K07072, K07144, K09733, K11212, K11780, K11781, K14941, K16792, K16793, and K18933. This pattern is an environment-specific signature of human gut methanogens and indicates a strong dependence on community-supplied intermediates. In contrast, rumen-derived methanogens were notably depleted in genes involved in the acetyl-CoA pathway genes, including K00192 (OR = 0.1), K00193 (OR = 0.08), and K00194 (OR = 0.06). The depletion of acetyl-CoA pathway genes in rumen methanogens indicates a reduced role for acetoclastic methanogenesis. This suggests that hydrogen is competitively diverted by coexisting rumen microbiome (e.g., acetogen) via the WLP, potentially limiting methane yield from acetate oxidation (Fig. 4).
From long-read metagenomic assemblies of rumen fluid, 67 high-quality MAGs were assigned to ecological roles based on methanogenesis-related gene content (Fig. 5). Genomes carrying genes that can supply substrates for methanogenesis were designated producers; archaeal genomes encoding the core methanogenesis machinery were designated methanogens; genomes encoding pathways that competitively consume methanogenesis substrates were designated competitors; genomes encoding methane oxidation were designated methanotrophs. These included 53 producers, 4 methanogens, 10 competitors, and 1 methanotroph.

Among competitor candidates, DNRA MAGs were identified as Bin.31__s_Aristaeella_sp900315675, Bin.35__s_UBA3792_sp002369195, Bin.38__s_UBA3792_sp902792295, Bin.39__s_Faecousia_sp900315595, Bin.41__s_Bulleidia_intestinalis, Bin.75__s_Denitrobacterium_detoxificans, Bin.122__s_Aristaeella_sp900322155, and Bin.126__s_Chordicoccus_sp900320575. The thermodynamically favorable nature of DNRA over methanogenesis positions these taxa as key electron sinks in methane-suppressing communities. DNRA offers a more favorable free-energy change than the reduction of CO2 to methane, enabling more efficient withdrawal of electrons. Rumen methane-mitigation strategies that supplement nitrate operate on this principle. The DNRA competitor candidates identified here support the feasibility of suppressing methanogenesis through electron competition [48]. The WLP was detected in Bin.4__g_FB2012 and Bin.73__s_Ruminococcus_sp002394695, suggesting these taxa divert hydrogen away from methanogenesis (Fig. 5). Given the depletion of acetoclastic methanogenesis in rumen methanogens, we prioritized taxa that can divert hydrogen as mitigation candidates, and the genus Ruminococcus appears to be a promising candidate for hydrogen competition in the rumen ecosystem.
DISCUSSION
This study includes two major components, an environment-based analysis of methanogenic archaea and a rumen shotgun MAG analysis. For the methanogen component, we combined established elements from prior workflows. The phylogenetic reconstruction followed the analysis flow of Ou et al. [49], and the pangenome analysis followed the framework of Prondzinsky et al. [11]. Environmental classification was assigned from the isolation context of each reference genome, allowing us to examine genetic features of methanogens alongside habitat-specific patterns. For the rumen MAG component, the assembly, mapping, and binning steps were guided by the Oxford Nanopore long-read shotgun workflow, which is well-suited for processing Oxford Nanopore Technologies data. Quality filtering and curation followed the methods of Richy et al. [50], and functional annotation was performed using eggNOG-mapper [20], consistent with the methanogen analysis. Although the rumen MAG pipeline is not directly derived from a single paper, once MAGs are recovered by binning, the subsequent interpretation is tool-independent, following the curation and QC sequence proposed by Richy et al. [50]. Similarly, eggNOG-mapper provides a complementary route to functional annotation in lineages, such as methanogens, where curated annotations remain limited. Therefore, these choices support the reproducibility of the workflow employed in this study.
Methanogenic archaea play a central role in regulating hydrogen flux and methane output across anaerobic ecosystems [1]. However, conventional reliance on cultivation-dependent methods and single-marker analyses has left substantial gaps in our understanding of their ecological differentiation and evolutionary dynamics [7]. To address this, we integrated broad phylogenetic analyses with genome-resolved comparisons to explore how environmental context shapes methanogen lineage structure and pathway composition, and how these patterns could inform methane-mitigation strategies [51]. The pangenome-based genomic analysis examines lineage-specific and niche-linked gene clusters that are involved in microbial metabolism, particularly methanogenesis, thereby identifying candidate genomic and metabolic targets for methane mitigation [52]. In line with this perspective, a methanogen pangenome analysis resolved a large conserved core together with habitat relevant accessory variability, providing a rational map to distinguish conserved enzymatic nodes from context-specific accessory modules that are pertinent to mitigation [11]. Advancing high-quality genome references and refining functional annotations will connect genotype to mechanism in rumen systems, supporting the design of targeted, microbiome-informed strategies to reduce methane emissions in livestock production [8].
The mcrA is a canonical component of methanogenesis and has served as a defining molecular marker for methanogens. Earlier studies suggested that mcrA sequences could substitute for 16S rRNA in taxonomic classification because both markers produced largely congruent phylogenetic trees [15,53]. However, more recent work has revealed substantial discordance between mcrA- and 16S-based phylogenies, influenced by environmental niche, taxonomic scope, primer design, and analytical framework [54]. These findings underscore the limitations of using any single functional gene as a universal proxy for evolutionary inference [55,56]. In other words, using a single functional gene as a representative marker to describe evolutionary relationships or to conduct phylogenetic analysis among different organisms can be inappropriate. Our results strongly reinforce this point: except for two minor clades (one comprising 5 methanogens from sewage and freshwater, and another comprising 6 methanogens from freshwater and seawater), no meaningful congruence was observed between 16S rRNA and mcrA trees (Fig. 2). This limited overlap suggests that phylogenetic resolution in methanogens operates primarily at the genus level and that evolutionary patterns are not clearly partitioned by habitat. The divergence reflects the conservative evolutionary trajectory of 16S rRNA compared with the more dynamic mcrA, which is subject to lateral gene transfer, modular pathway organization, and selective gene loss [57,58]. Supporting this, functional analyses revealed that methanogens exhibited low retention of acetoclastic methanogenesis genes, whereas human gut methanogens showed marked depletion in cofactor biosynthesis modules for coenzymes M, B, F420, and methanofuran (Fig. 4). These pathway differences argue against a single canonical methanogenesis route across habitats and help explain why mcrA-based phylogeny alone does not recapitulate 16S rRNA relationships. Therefore, the phylogenetic and genome content results support a marker strategy that integrates 16S rRNA with multiple pathway genes and genome-resolved context, rather than relying on mcrA alone, for robust taxonomic inference and for ecological interpretation across environments.
Because mcrA did not reliably capture environment-specific structure in our phylogenies, we integrated pangenome-based clade definitions with environmental metadata to track niche-linked genomic adaptations. The methanogen pangenome revealed a clear hierarchical organization, with core and soft-core gene sets enriched in housekeeping functions such as translation (COG J), amino acid metabolism (COG E), and coenzyme metabolism (COG H; Fig. 3A). This pattern is in accordance with previous archaeal pangenomic studies that reported similar enrichment of informational processes in conserved gene sets [59]. The archaeal distribution contrasts with typical bacteria, where J, E and H tend to occur alongside C and F for energy metabolism and nucleotide transport and metabolism at comparable proportions [45]. These results indicate strong conservation of transcription, translation, and replication in archaea, and also suggest a higher fraction of proteins lacking confident annotation due to limited study coverage [44,46]. The shell gene set further contained numerous poorly annotated or lineage-specific orthogroups, highlighting persistent annotation gaps within archaeal genomics. Within the rumen-derived methanogen clade, the shell showed enrichment for cell wall, membrane, and envelope biogenesis, consistent with tight attachment to symbiotic bacteria in the rumen and with sustained requirements for maintenance and remodeling of archaeal cell envelopes based on pseudomurein and S-layers (Fig. 3B) [60]. This interpretation aligns with prior observations of Methanobrevibacter ruminantium M1, which encodes multiple adhesin-like proteins such as Mru_1499 capable of binding directly to protozoa and hydrogen-donating bacteria [61]. The ether lipid membrane typical of archaea also requires compositional maintenance under volatile fatty acids, long-chain fatty acids, and osmotic stress prevalent in the rumen, which helps explain the relative expansion of membrane and envelope biosynthesis pathways [47]. The observed depletion of COG J outside the core aligns with the broader archaeal trend, which preserves translation and ribosome biogenesis functions as deeply conserved core features [62].
Methanogenesis is generally categorized into hydrogenotrophic, acetoclastic, and methylotrophic pathways [63]. Our KEGG-based screening revealed that genes supporting methylotrophic methanogenesis were retained at relatively low frequencies (mean ratio 0.32) across environments (Fig. 4), consistent with previous reports indicating that hydrogenotrophic methanogenesis is the dominant route, with methylotrophy operating as a niche-specific, facultative strategy [64]. The result reflects gene presence only, and previous studies show that methylotrophic methanogenesis can become prominent in settings with abundant methylated substrates [1,65]. Among environment-specific distinctions, human gut methanogens exhibited pronounced depletion of cofactor biosynthetic genes associated with coenzymes M, B, F420, and methanofuran. This is consistent with metabolic streamlining during host-associated adaptation [66]. Human gut-associated Methanomassiliicoccales exemplify this trend, with loss of canonical coenzyme M biosynthesis genes and atypical energy conservation modules, as well as loss of the Wood–Ljungdahl methyl branch, supporting a hydrogen-dependent methyl-reducing lifestyle that increases reliance on community-supplied intermediates [67,68]. Alternative or yet-unresolved routes for coenzyme M formation and exogenous supply have been proposed, which together suggest that human gut methanogens may depend more on community interdependence and pathway substitution than on strict de novo cofactor synthesis [66,69].
Interestingly, depletion of acetyl-CoA pathway genes was clearly observed in rumen-derived methanogens (Fig. 4). This pattern supports that hydrogenotrophic methanogenesis predominates in the rumen and that acetoclastic activity is low [70]. It consists of the dominance of Methanobrevibacter, which relies primarily on the hydrogenotrophic route, and the low prevalence of Methanosarcinales, which can perform acetoclastic methanogenesis. Together, these features indicate limited acetate use in rumen methanogenesis. The deficit of acetyl-CoA pathway genes in rumen methanogens is therefore best interpreted as an outcome of ecological selection pressures [71,72]. Acetate has been considered a substrate for acetoclastic methanogenesis, which led to the view that acetate-forming bacteria hinder methane mitigation [73]. Recent studies instead highlight competition for metabolic hydrogen as the main control [74,75]. When methanogenesis is suppressed, electron flow is redirected toward alternative hydrogen sinks such as propionate formation and reductive acetogenesis, with accompanying increases in volatile fatty acid yields. Consistent with this mechanism, recent genome-resolved and metabolite-profiling work shows that 3-nitrooxypropanol (3-NOP) cuts methane by ~60% while stimulating reductive acetogenesis and shifting SCFA and H2 dynamics without depressing intake [76]. A previous meta-analysis also indicated consistent shifts toward propionate and activation of acetogenic lineages, while acetoclastic methanogenesis appears uncommon in the rumen [77]. Taken together, these results support positioning acetate production as part of a reallocation of hydrogen sinks among the competitive routes that can antagonize methanogenesis under inhibition regimes [78].
In the genome-resolved functional classification based on rumen shotgun sequencing data, competitors were divided into a DNRA group that draws electrons away from methanogenesis and a WLP group that competes for H2. Thermodynamically, DNRA offers a larger free-energy gain than CO2 reduction. Under standard conditions, the reduction of CO2 to CH4 is on the order of –131 kJ mol–1, whereas the reduction of NO3– to NH3 is on the order of –600 kJ mol-1, making DNRA a stronger electron sink [79]. This thermodynamic advantage intensifies competition for the reductants used in methanogenesis, and nitrate has been widely evaluated as a rumen methane mitigation agent [80]. Recent in vitro and applied studies corroborate nitrate-driven methane suppression with concomitant microbiome and fermentation shifts, including dose-dependent responses and cation-specific effects [81]. Prior studies reported methane reductions with Denitrobacterium supplementation alone, in combination with nitrate, and with the combined treatment showing a greater effect than either alone, consistent with electron diversion via DNRA by Denitrobacterium [48,82]. Consistent with this, our analysis detected DNRA genes in genera such as Aristaeella, Faecousia, Bulleidia, and Chordicoccus (Fig. 5). Although direct evidence linking these genera to DNRA remains limited and precludes immediate designation as nitrate reducers, their gene content highlights them as competitor candidates for future methane mitigation assays. In the rumen, the WLP is thermodynamically and kinetically disadvantaged while methanogenesis dominates, yet it can emerge as an alternative H2 sink when methanogenesis is suppressed or when H2 partial pressure increases [83]. Classical incubations and experiments with methanogenesis inhibitors such as BES repeatedly showed activation of indigenous acetogens and increased acetate production under methanogenesis suppression, supporting an inhibition-induced shift toward WLP [84,85]. More recent meta-omics studies indicate the presence of acetogenic lineages not only within Lachnospiraceae but also within Ruminococcaceae, with enrichment of WLP marker genes and acetate formation when methanogenic pressure is reduced [86,87]. In the present study, the detection of Ruminococcaceae MAGs including fhs and cooS as WLP markers suggests potential facultative switching to an H2-sink role, even though these taxa are generally recognized as primary fermenters that produce H2 (Fig. 5). Unlike DNRA, WLP does not depend on an external electron acceptors, but under localized H2 accumulation or methanogenesis inhibition, it can operate as a competing electron sink and reroute H2 flow in ways that contribute to methane mitigation [88].
Our findings confirm that the comparative genome-resolved approach is a powerful tool for identifying a targeted strategy to mitigate ruminant methane production. Integrating phylogenetic and pangenome information allows the approach to focus on a practical set of organisms and modules for experimental validation. The WLP, identified as a novel candidate, provides a concrete establishment for targeted assays and follow-up experiments. These findings enhance our genome-resolved understanding of methanogens and reveal how their metabolic pathways vary in response to ecological niche differentiation. However, functional annotation of archaeal genomes remains incomplete because there are still uncharacterized proteins compared to bacteria, which limits pathway-level inference and experimental validation due to the limited cultivability of methanogens. Coordinated multiomics workflows, integrating expanded archaeal genome references with culturomics, metatranscriptomics, and metabolomics, will be essential to confirm whether the predicted roles are expressed under actual rumen conditions. Systematic curation of archaeal functional databases and targeted validation of key modules highlighted by pangenome analyses may enhance annotation precision and strengthen the mechanistic relation between methanogen genomics and methane-mitigation strategies, thereby supporting the development of more effective and sustainable interventions in livestock production.
SUMMARY AND CONCLUSION
In this study, we integrated broad phylogenetic reconstruction with pangenome-resolved analyses to interrogate the diversity and functional attributes of methanogens across diverse ecosystems. By constructing both 16S rRNA- and mcrA-based phylogenies and coupling them with pangenome comparisons, we assessed habitat-specific clustering and identified gene-level signatures associated with environmental adaptation. Using rumen shotgun sequencing metagenomes, we determined putative methanogenic producers, hydrogen competitors, and methanotrophs within the methane network and prioritized candidate taxa for in vitro methane mitigation strategies. In contrast, rumen methanogens are enriched for genes linked to cell envelope biogenesis, consistent with sustained physical interactions in the rumen. Furthermore, the observed depletion of acetyl-CoA pathway genes in rumen methanogens suggests limited acetoclastic activity and raises the potential for hydrogen redirection via the WLP in non-archaeal partners. These findings support a functional marker strategy that integrates 16S rRNA with pathway-specific genes and a pangenome framework to enhance ecological interpretations of methanogens and to prioritize potential targets for methane mitigation in ruminants.