
INTRODUCTION
According to assessment reports by the Intergovernmental Panel on Climate Change (IPCC), the increasing greenhouse gas emissions are leading to an increase in the rise of global mean temperatures, from 1.8°C in 1980–1999 to 4.1°C in 2090–2099 based on the climate models [1]. Because livestock systems are greatly affected by environmental conditions, livestock production is under increasing pressure from climate change due to it causing heat stress in the animals [2]. Heat stress is an environmental factor that negatively affects cattle by reducing their feed intake, which ultimately decreases their metabolic heat production and productivity [3]. Heat stress also alters carbohydrate, lipid, and protein metabolism irrespective of feed intake reduction [4]. However, susceptibility to heat stress varies among animals due to differences in behavior, adaptability, diet, productivity level, radiant heat load, genotype, and the color and thickness of the hair coat [3].
The Hanwoo breed is a taurine type of cattle that is native to Korea. It has a small body size and, since the 1960s, has been improved as beef cattle due to its high palatability and desired chewiness [5]. Hanwoo is the most dominant breed of cattle and its populations account for more than 90% of the total beef cattle in Korea [6]. Therefore, it is important to assess the vulnerability and adaptability of Hanwoo cattle to heat stress, especially in light of predicted climate change scenarios.
The rumen microbiome plays an important role in the digestion of plant materials and rumen fermentation in ruminants, and this suggests that optimizing rumen fermentation will lead to improvements in nutrient utilization and productivity of cattle [7]. The composition of the rumen microbiome is affected by the host breed, host age, diet, season, and geographic region [7]. To the best of our knowledge, only two previous studies have assessed the impact of heat stress on the rumen microbiome composition of Holstein heifers, wherein significant shifts were observed [8,9]. However, since then, no additional studies have reported on the effects of heat and humidity on the rumen microbiome and the impact of heat stress on the rumen microbiome of Hanwoo cattle has not been assessed. It is important to determine the adaptability of Hanwoo cattle under heat stress thus, in this study, we evaluated the impact of heat stress in humid conditions on the composition of the rumen microbiome of Hanwoo cattle using next-generation sequencing.
MATERIALS AND METHODS
All experimental procedures were reviewed and approved by the Institutional Animal Care and Use Committee of the National Institute of Animal Science, Korea (No. 2015-134).
Four Hanwoo steers (body weight = 234.5 ± 24.0 kg) that were 12 months of age were individually housed in climate-controlled chambers with 60% humidity and fed a diet consisting of 60% concentrate and 40% rice straw. The climate-controlled chambers were maintained at 15°C (environmental temperature) for 2 weeks, after which four rumen samples were collected from each of the four Hanwoo steers (0-day group) using the stomach tubing method, as previously described [10]. The environmental temperature was then raised to 35°C for 6 days. Four rumen samples were collected from the four Hanwoo steers after 3 days (3-day group) and after 6 days (6-day group). All 12 rumen samples (4 animals × 3 sampling periods) were stored at −80°C until extraction of the total community DNA.
The total community DNA was extracted from the 12 rumen samples using the repeated bead beating plus column (RBB+C) method [11]. The V3-V4 region of the bacterial 16S rRNA gene was amplified using the 341F (5’-CCTACGGGNGGCWGCAG) and 805R (5’-GACTACHVGGGTATCTAATCC) primers. The V1-V3 region of the archaeal 16S rRNA gene was amplified using the Met86F (GCTCAGTAACACGTGG) and Met471R (GWRTTACCGCGGCKGCTG) primers. The resulting amplicon libraries were sequenced using the Illumina MiSeq platform (Illumina, San Diego, CA, USA). After sequences were assembled using the FLASH program [12], all sequencing processes were conducted using the QIIME 1.9.1 software package [13]. Chimeric sequences were identified using the ChimeraSlayer program [14]. The resultant non-chimeric sequences were assigned to taxa with reference to the Greengenes database [15] while the operational taxonomic units (OTUs) were determined at a 97% sequence similarity using the uclust program [16]. In total, 40,000 sequences were subsampled to normalize the number of OTUs from each sample and then used to analyze the alpha diversity. The beta diversity was analyzed using the principal coordinate analysis (PCoA) plot on the unweighted UniFrac distance matrix.
The relative abundance of the total sequence reads for each taxon and the alpha diversity indices were compared among the three sample groups with an ANOVA followed by Duncan’s multiple comparisons with XLSTAT statistical software (Addinsoft, New York, NY, USA). Significant differences were determined at p-values < 0.05.
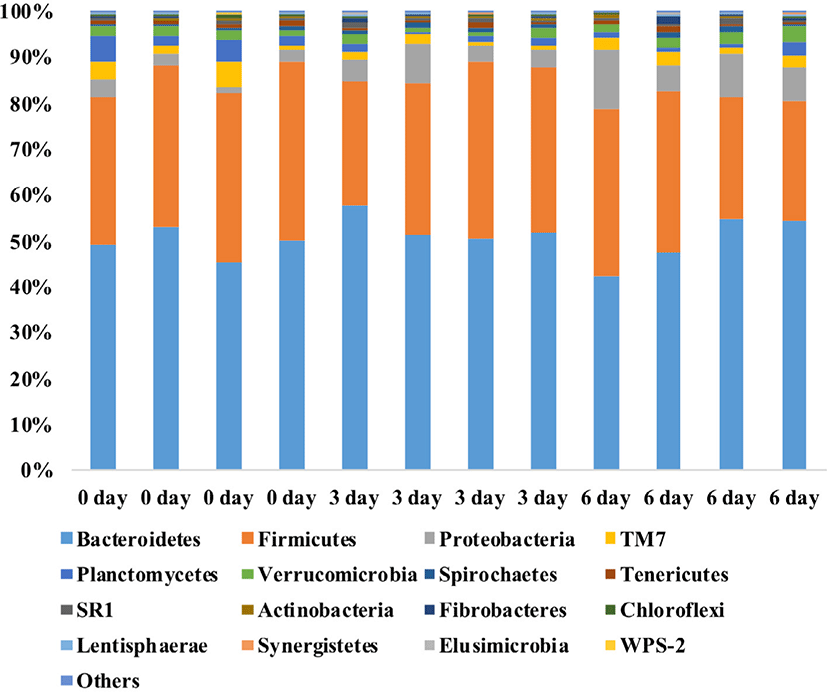
RESULTS
A total of 997,843 bacterial sequences were obtained from the 12 rumen samples. Taxa representing > 0.1% of the total sequence reads on average were regarded as the “major taxa”, and their relative abundances were statistically compared among the three groups (0-day, 3-day, and 6-day). Bacteroidetes was the most dominant phylum with individual samples accounting for 42.3% to 58.0% of the total sequences, followed by Firmicutes (26.2%–39.0%), Proteobacteria (1.4%–12.9%), TM7 (0.9%–5.2%), Planctomycetes (0.5%–5.5%), Verrucomicrobia (0.7%–3.3%), Spirochaetes (0.3%–1.5%), Tenericutes (0.4%–1.4%), SR1 (0.2%–1.0%), Fibrobacteres (0.0%–1.4%), Actinobacteria (0.1%–0.7%), Chloroflexi (0.1%–0.9%), Lentisphaerae (0.1%–0.7%), Synergistetes (0.0%–0.4%), Elusimicrobia (0.0%–0.3%), and WPS-2 (0.0%–0.2%) (Fig. 1). The relative abundances of these 15 phyla did not differ (p > 0.05) among the three sampling groups, except Proteobacteria, Planctomycetes, and Chloroflexi. The relative abundances of Proteobacteria and Chloroflexi were lower (p < 0.05) in the 6-day group than in the 0-day group, while the relative abundance of Planctomycetes was lower (p < 0.05) in the 3-day group than in the 0-day group.
The Bacteroidetes sequences were assigned to eight major families, including Prevotellaceae (10.2%–29.8%), S24-7 (0.9%–12.6%), Paraprevotellaceae (3.6%–10.0%), BS11 (0.3%–3.5%), Porphyromonadaceae (0.0%–3.3%), RF16 (0.1%–0.9%), Bacteroidaceae (0.0%–0.8%), and Weeksellaceae (0.0%–0.3%). Relative abundances of these eight major families did not differ (p > 0.05) among the three sampling groups except for Prevotellaceae, which was greater (p < 0.05) in the 6-day group than in the other two groups (Fig. 2A). Five major genera of Bacteroidetes were identified, including Prevotella (7.7%–31.7%), CF231 (1.8%–5.2%), YRC22 (0.7%–2.7%), Paludibacter (0.0%–2.5%), and BF311 (0.0%–0.8%). Relative abundances of these five major genera did not differ (p > 0.05) among the three sampling groups except for Prevotella and YRC22, which were the most abundant (p < 0.05) in the 6-day group among the three sampling groups (Fig. 3A).
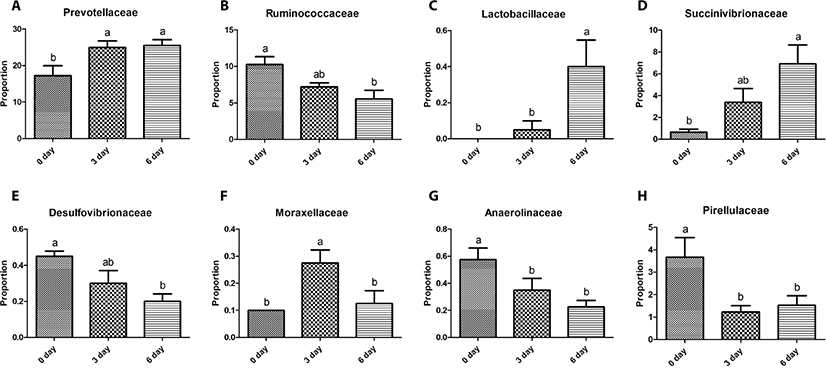
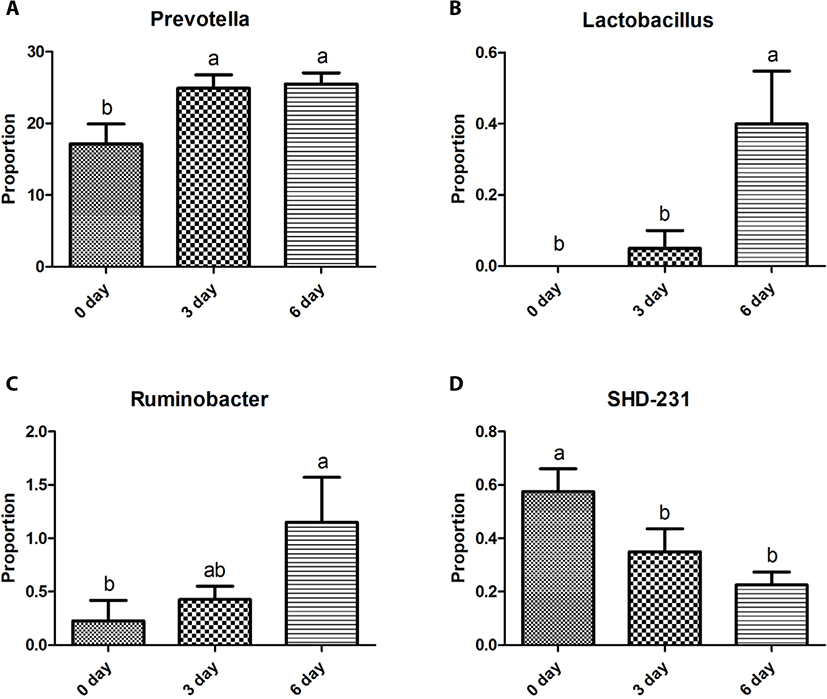
The Firmicutes sequences were assigned to nine major families, including Ruminococcaceae (3.7%–12.2%), Veillonellaceae (5.6%–14.4%), Lachnospiraceae (2.7%–8.0%), Erysipelotrichaceae (0.5%–3.5%), Clostridiaceae (0.3%–1.0%), Christensenellaceae (0.2%–2.4%), Mogibacteriaceae (0.2%–1.5%), and Lactobacillaceae (0.0%–0.7%). Relative abundances of these nine major families did not differ (p > 0.05) among the three sampling groups, except for Ruminococcaceae and Lactobacillaceae. The relative abundance of Ruminococcaceae was lower (p < 0.05) in the 6-day group than in the 0-day group, whereas that of Lactobacillaceae was greater (p < 0.05) in the 6-day group than in the other two groups (Figs. 2B and C). Twelve major genera of Firmicutes were identified, including Succiniclasticum (2.1%–4.9%), Ruminococcus (0.4%–7.5%), Butyrivibrio (0.5%–3.0%), Selenomonas (0.3%–2.9%), Anaerovibrio (0.1%–3.2%), RFN20 (0.2–1.7%), Clostridium (0.1%–1.1%), Pseudobutyrivibrio (0.1%–1.0%), Coprococcus (0.0%–1.4%), p-75-a5 (0.1%–0.4%), Lactobacillus (0.0%–1.0%), L7A_E11 (0.0%–0.8%), and Moryella (0.0%–0.4%). Relative abundances of these genera did not differ (p > 0.05) among the three sampling groups except for Lactobacillus, which was greater (p < 0.05) in the 6-day group than in the other two groups (Fig. 3B).
The Proteobacteria sequences were assigned to four major families, including Succinivibrionaceae (0.2%–11.0%), Desulfovibrionaceae (0.1%–0.5%), Moraxellaceae (0.0%–0.4%), and Pasteurellaceae (0.0%–0.3%), with significant differences (p < 0.05) among the three sampling groups. The relative abundance of Succinivibrionaceae was greater (p < 0.05) in the 6-day group than in the 0-day group, whereas that of Desulfovibrionaceae was lower (p < 0.05) in the 6-day than in the 0-day group (Figs. 2D and E). The relative abundance of Moraxellaceae was greater (p < 0.05) in the 3-day group than in the other three groups (Fig. 2F). Three major genera of Proteobacteria were identified, including Desulfovibrio (0.1%–0.4%), Ruminobacter (0.0%–2.1%), and Succinivibrio (0.1%–5.5%). Relative abundances of these 3 genera did not differ (p > 0.05) among the three sampling time groups except for Ruminobacter, which was greater (p < 0.05) at the 6-day group than at the 0-day group (Fig. 3C).
The remaining 11 major phyla identified were assigned to 16 major families Bifidobacteriaceae (0.0%–0.5%), Coriobacteriaceae (0.1%–0.4%), Anaerolineaceae (0.1%–0.8%), Elusimicrobiaceae (0.0%–0.3%), Fibrobacteraceae (0.0%–1.4%), Victivallaceae (0.1%–0.6%), Pirellulaceae (0.5%–5.5%), Sphaerochaetaceae (0.0%–0.6%), Spirochaetaceae (0.2%–0.9%), Dethiosulfovibrionaceae (0.0%–0.3%), Anaeroplasmataceae (0.0%–0.3%), Mycoplasmataceae (0.0%–0.9%), F16 (0.9%–5.2%), R4-41B (0.0%–0.5%), RFP12 (0.5%–2.1%), and WCHB1-25 (0.0%–0.3%) with no difference among the three sampling groups, except for Anaerolineaceae and Pirellulaceae. Relative abundance of Anaerolineaceae was greater (p < 0.05) in the 0-day group than in the 6-day group, while that of Pirellulaceae was greater (p < 0.05) in the 0-day group than in the 3-day group (Figs. 2G and H). Four major genera of the eleven phyla were identified, including Fibrobacter (0.0%–1.6%), Sphaerochaeta (0.0%–0.6%), and Treponema (0.1%–0.9%), and SHD-231 (0.1%–0.6%). Relative abundances of these four genera did not differ among the three sampling groups except for SHD-231, which was greater (p < 0.05) in the 0-day group than in the 6-day group (Fig. 3D).
A total of 1,508,770 archaeal sequences were obtained from 12 rumen samples and assigned to phylum Euryarchaeota. The Euryarchaeota sequences were assigned to two major families, including Methanobacteriaceae (90.5%–99.5%) in class Methanobacteria and Methanomassiliicoccaceae (0.5%–9.4%) in class Thermoplasmata. Relative abundances of these two major families did not differ (p > 0.05) among the three sampling groups. Two major genera of the family Methanobacteriaceae were identified, including Methanobrevibacter (89.4%–99.3%) and Methanosphaera (0.2%–2.2%), while one major genus, vadinCA11 (0.5%–9.4%), of family Methanomassiliicoccaceae was identified. Relative abundances of these three genera did not differ (p > 0.05) among the three sampling groups.
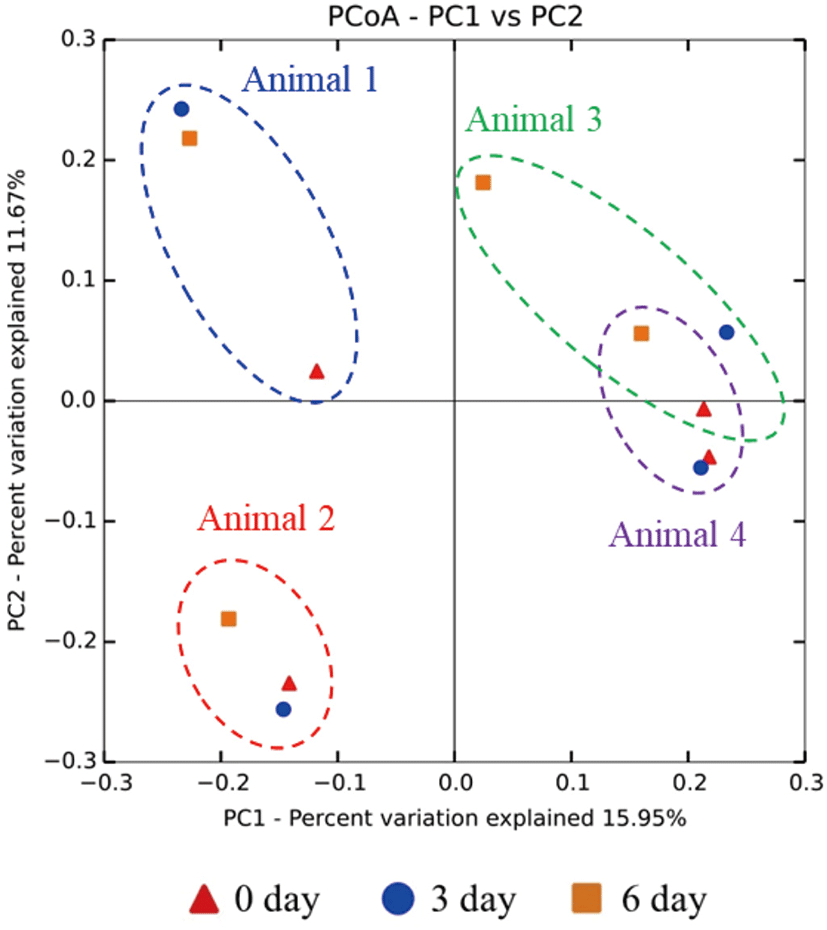
From each rumen sample, 40,000 bacterial (40,000 sequences × 12 samples = total 480,000 bacterial sequences) and 40,000 archaeal (40,000 sequences × 12 samples = total 480,000 archaeal sequences) sequences were randomly sub-sampled for normalization of the number of OTUs. The alpha diversity indices (OTU richness, Chao1, PD_whole_tree distance, Shannon diversity index, and Simpson diversity index) for both the bacteria and archaea did not differ (p > 0.05) among the three sampling groups (Table 1). In the beta diversity PCoA plot, individual microbiomes of the four study animals were separated. The rumen microbiome of the 6-day group was distinct from the rumen microbiomes of the other two groups in each study animal (Fig. 4). These results indicated that the composition of the rumen microbiome not only differs among individual animals but is also affected by the period of exposure to heat stress.
2) From each rumen sample, 40,000 sequences were randomly subsampled to normalize the number of OTUs.
DISCUSSION
To date, few studies have been conducted to assess the impact of temperature and humidity on the composition of the rumen microbiome in cattle. Tajima et al. [8] examined the effect of heat stress on rumen microbial diversity in Holstein heifers. However, to our knowledge there are no reports on the composition of the rumen contents of Hanwoo cattle, a Korean native breed. We believe that the present study is the first to investigate both the ruminal bacterial and archaeal diversity in Hanwoo cattle under heat stress.
Although previous studies identified the changes in some ruminal bacterial species in response to increased temperature and humidity, the composition of ruminal bacterial communities was evaluated using traditional cloning and Sanger sequencing [8,9]. Because of the limitation of the clone library method, only hundreds of sequences were able to be analyzed with a low percent coverage at the species level. Because a small number of sequences may result in poor quantitative repeatability [17,18], the use of cloning and the Sanger sequencing method with small sequence reads may lead to biased results. The present study, however, analyzed more than 15,000 sequence reads per sample using next-generation sequencing and evaluated the composition of the rumen microbiome with a very high percent coverage.
Normal rumen microbiome in Hanwoo cattle showed disruption at 6 days in response to heat stress, i.e., a decrease in fibrolytic Ruminococcaceae and an increase in lactate-producing Lactobacillaceae. The low pH resulting from the lactate produced by the lactate-producing bacteria may inhibit fiber-attachment of ruminal fibrolytic bacteria, resulting in the decrease in fiber digestibility [19]. The putative genus SHD-231 was identified as bacteria that adhere to ingested forage in a previous study [20]. Therefore, SHD-231 may be an unknown fibrolytic bacterial genus present in the rumen. In this study, decreased SHD-231 in the 6-day group may be due to low pH, which reduces bacterial attachment for fiber degradation [21]. The abundance of the fibrolytic Prevotella strains that are sensitive to low pH were decreased whereas the amylolytic Prevotella strains that are tolerant against low pHs were increased in the 6-day group [22]. Amylolytic Ruminobacter also increased under the risk of rumen acidosis in response to heat stress as described previously [23]. The bacteria in the putative genus YRC22 in Paraprevotellaceae may also be acid-tolerant, which resulted in its observed increase in the 6-day group. Because Desulfovibrio can ferment lactate [24], the increase in Desulfovibrio in the 6-day group may have been a result of the utilization of the lactate produced in response to heat stress.
Methanobrevibacter greatly contributes to the methane emissions irrespective of the ruminant species and breeds [7]. In the Hanwoo cattle, Methanobrevibacter was the most dominant genus. Because rumen methanogens are not significantly altered under heat stress, methane emissions from Hanwoo cattle may not be changed by short-term exposure to heat stress. However, methane emissions from Hanwoo steers will need to be directly measured in future studies to confirm if they are affected by short-term or long-term heat stress.
Short-term heat stress did not quickly alter both the archaeal and bacterial diversity. Although the cellulolytic and lactate-producing bacteria were affected by short-term heat stress, most of the ruminal microbes were stable and were not affected. However, rumen microbial diversity might be altered by long-term heat stress. Time points that result in shifts in rumen microbial diversity in Hanwoo cattle will need to be determined in future studies. The understanding of such time points may contribute to the development of different feeding strategies to extend these time points and alleviating the suffering of cattle under heat stress.
The composition of rumen bacterial diversity is affected by different breeds having different host genetics [25]. Therefore, the adaptability of Hanwoo cattle to high-temperature and high-humidity conditions may be related to host genetics that influence stable bacterial diversity in the rumen. Future studies will need to examine the impact of the host on the ruminal bacterial diversity in Hanwoo cattle to further develop strategies to reduce heat stress in Hanwoo cattle.
In conclusion, this is the first study to demonstrate that heat stress from short-term exposure to high temperature conditions can affect the composition of the rumen microbiome of Hanwoo steers. Six days of heat stress resulted in a decrease in fibrolytic bacteria that are sensitive to low pH conditions and a decrease in amylolytic bacteria that are resistant to low pH conditions, which indicated that risk of lactic acidosis in the rumen can be increased after 6 days under high temperature conditions. In future studies, the ruminal microbial shifts in response to long-term heat stress will need to be examined to further our understanding of the risk of ruminal acidosis and to develop strategies to prevent ruminal acidosis under heat stress in Hanwoo cattle. Since the physiology of Hanwoo cattle differs from that of other cattle breeds, this study may provide valuable data with which to further assess the adaptability of Hanwoo cattle and different feeding strategies to restore rumen microbiomes that have been disrupted by heat stress.