
Background
Purine nucleoside xanthosine (XS) when added to asymmetrically dividing stem cells, induceds a transition to symmetrical division, which ultimately leads proliferation of stem cells. Suppression of asymmetrical division of cells by XS is regulated through inosine monophosphate dehydrogenase (IMPDH) in p53 dependant fashion [1]. XS is converted into xanthosine 5′-monophosphate (XMP) by the enzyme IMPDH. Thus, XS has the essential role in IMPDH regulation. The intramammary infusion of xanthosine (XS) into lactating cow, has been shown to increase mammary stem cell population and has been hypothesized to increase milk production [2]. Contrarily, xanthosine treatment shown to have no effect on mammary stem cell population [3]. Therefore, to make a distinct and clear effects of XS on mammary gland, more research are warranted.
Milk is secreted by the mammary epithelial cells (MECs), which are gradually exfoliated during lactation. Because of the gradual exfoliation and constant replenishment of these MECs, it is likely that gene expression differences in these cells may be major contributors to the overall process of lactation. Several methodologies have been developed to isolate and enrich MECs from mammary glands for transcriptional profiling of MEC. One methodology involves the surgical biopsy of mammary tissue followed by the preparation of singular cell suspension and in vitro propagation of MECs in culture [4]. Alternatively, immunomagnetic methods of MEC cell seperatation from milk somatic cells have also been attempted by several studies [5, 6]. Recently, non-invasive approaches of extracting MECs from milk have been developed that involve the capture of milk fat globules (MFG) from the milk fat layer. These MFGs contain MEC cytoplasmic remnants, called crescents, due to their moon shaped appearance under the light microscopy [7]. The percentage of MFG crescents in milk varied among the mammalian species with a high percent volume (3–8%) in human milk to a low percent volume (< 1%) in cattle [8, 9]. MFG crescents, being its cytoplasmic origin, are a source of MEC-specific RNA. In the analysis of the human MFG transcriptome using microarrays and RNA-sequencing, it has been confirmed that the MFG transcriptome include genes uniquely expressed in the MEC [7, 10, 11]. These studies have established MFGs as a reliable source for MEC-specific gene expression analysis during lactation.
The main aim of the present study was to identify XS-induced, gene expression differences in MECs by deep sequencing of the total RNA associated with MFG during early lactation in Beetal goats. Using RNA-sequencing, we sought to identify the differentially expressed genes (DEGs) between XS treated (TRT) and control glands (CON). Additionally, using reverse transcriptase quantitative PCR (RT-qPCR), we sought to confirm observed differential gene expression and fully characterized the transcript abundance of genes of interest.
Methods
Use of goats for this study was approved by the “Committee for the Purpose of Control and Supervision of Experiments on Animals” (CPCSEA; reference no. 25/20/2016), Ministry of Environment, Forest and Climate Change (Animal Welfare Division), New Delhi. Eight primiparous Beetal goats were used in this experiment. A 20 mL suspension of 10 mM XS was infused through the teat canal and 1 mL of intra-parenchymal injection was given to one randomly chosen mammary gland (half udder) for 3 consecutive days, twice daily (once in the morning, and once in the evening immediately after milking). Complete milk removal was performed for each TRT glands, followed by the insertion of a sterile 20 gauge blunt needle into the teat canal to extract all of the remaining milk secretion. The XS powder was diluted in sterile saline and sterilized with a 0.22 μm filter before infusions. To ensure the delivery of XS into the peripheral regions of the glands, an additional injection of 1 mL XS was administered intraparenchymally according to the methods used in a similar study [2]. For each goat, one mammary gland was treated with XS while the other gland was used as control (CON) and was not infused with XS. One week after the last XS injection, milk was collected for RNA isolation from both the TRT and CON glands of each animal.
Freshly collected milk were mixed with 0.1 0.1% acridine orange (AO) and incubated for 5 min at room temperature to stain nucleic acids (DNA and RNA). A 10 μL of AO-stained milk were placed onto positive charged slides and covered with a coverslip which was then sealed with nail polish to prevent milk evaporation. Slides were viewed immediately on an inverted fluorescent microscope (Nikon Eclipse 90i, Tokyo, Japan). Two images were captured for each slide, a red fluorescence channel TRITC for AO-intercalated RNA, and a green fluorescence channel FITC to view AO-intercalated DNA. AO-stained milk Images were analyzed using Fiji as described earlier [8]. The accuracy of this method was confirmed manually by comparing a subset of processed and original images. Overlays of the AO- RNA channel (Red) with the AO-DNA channel (green) images were analysed using Fiji [12] (Fig. 1).

Total RNA was extracted using a combination of Trizol (Invitrogen, Carlsbad, CA, USA) and GenElute Mammalian Total RNA isolation kit (Sigma, St. Louis, MO, USA) as published earlier [13]. Briefly, for each 40 mL milk sample after centrifugation, 500–700 μg of milk fat was collected using a 1 mL sterile microtip, and put into a 5 mL nuclease free tube (Genaxy Scientific Pvt. Ltd. New Delhi, India) containing 1.5 mL of Trizol™ reagent (ThermoFisher Scientific, Waltham, USA). The milk fat was homogenized in Trizol for 2–3 min by vortexing. The homogenized mixture was incubated at room temperature (RT) for 3 min and stored at − 80 °C for further use. The homogenates were thawed quickly by rubbing with palm, vortexed briefly and centrifuged at 12000 x g for 5 min at 4 °C to remove excess fat (on top). Clean homogenate (mid layer) was transferred into a new, 2 mL RNAase-free tube, containing 300 μL of chloroform (HiMedia Laboratories Pvt. Ltd. Mumbai, India). After vigorous shaking for 30 s, the mixture was incubated at RT for 10 min for complete solubilization of fat. Tubes were centrifuged at 12000 x g for 15 min at 4 °C. A 300 μL of the clear supernatant was used for total RNA extraction using the GenElute Mammalian Total RNA isolation kit, following the manufacturer’s instructions. RNA quantity was measured using a NanoDrop 1000 (ThermoFiesher Scientific, Waltham, USA). RNA integrity was determined using a Tapestation (Agilent Technologies, Santa Clara, USA) before the samples were processed for RNA sequencing.
Library preparation, quality control, sequencing and data analysis were performed at a commercial sequencing facilty (SciGenome, Kochi, India). Due to budget constraints and feasibility of RNA sequencing of milk fat derived RNA, only four libraries (two XS treated- 741 L and 647 L and two control- 741R and 647R) were considered from two individual goats. To generate each library, 5–10 μg of total RNA was isolated and prepared using a TruSeq RNA sample preparation kit V2 (Cat:RS-122-2001, Illumina, San Diego, CA, USA). Size distribution of the sequencing library was determined by gel electrophoresis. Library quantification was done using Qubit 3.0 fluorometric quantitation system (ThermoFisher Scientific, Waltham, USA). Paired-end sequencing was performed on Illumina’s HiSeq 2500 platform. Approximately 40 million paired-end, 2 × 100 bp reads were generated for each sample. Raw fastq files have been submitted to NCBI Sequence Read Archive (SRA) database with the study accession PRJNA389156 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA389156/).
Initial checking of raw sequences for the quality has been performed by the sequencing vendor. Briefly, reads containing adaptors were filtered from the dataset. To avoid base composition bias, 12 bases were trimmed at the 5′ end of both read pairs. RNA sequencing read quality checks included base quality score distribution, sequence quality score distribution, average base content per read, GC distribution in the read and occurrence of over-represented sequences. Raw reads were filtered when the average of their base quality scores was less than 20. Processed reads were aligned to the goat reference genome downloaded from NCBI (GCA_001704415.1_ARS1) [14]. Sequencing reads from non-coding RNAs were excluded from the analysis. Sequence alignment was performed using the STAR program (ver.2.5.2b). Two methods were used for differential gene expression analysis. First, the quantification of the raw expression of genes and transcripts was performed using the cufflinks program (ver. 2.2.1). The expression values were normalized to a FPKM (fragment per kilo per million) value for each of the genes and transcripts. Differential expression analysis was performed using cuffdiff program within the cufflinks package. Second, Deseq2 [15] was also used to identify significant differentially expressed genes. In this analysis, RUVseq [16] was used to remove any potential unwanted variation in gene quantification using several housekeeping genes. We compared the mRNA profile of TRT samples to that of the CON samples. A total of four mRNA sequence datasets (741 L, 741R, 647 L and 647R, as described in previous section) were used.
First-strand cDNA was synthesized from 1000 ng of total RNA using iScript Reverse Transcription Supermix (BioRad Laboratories, CA, USA) according to the manufacturer’s instructions. Resultant cDNA was stored at − 20 °C. The RNA isolated from MFG was used to quantify changes in gene expression by real time thermocycler (CFX96 Touch™ Real-Time PCR) using iTaq™ Universal SYBR Green Supermix (BioRad Lab. California, USA). Gene specific primers were designed on the basis of goat reference mRNA sequence obtained from NCBI using NCBI Primer-BLAST following guidelines described previously [17]. Desalted oligos (25 mM) were purchased from Integrated DNA Technologies (IDT Inc., Iowa, USA).
Each 10 μL of PCR reaction contained each 0.25 μL of forward and reverse primers (2.5 pmol), 5 μL of Brilliant III Ultra-Fast SYBR® QPCR (Agilent Technologies, Santa Clara, USA), 3 μL of cDNA (1:10 dilution) and 1.5 μL of nuclease free water. Two-step RT-qPCR assays were performed with the following cycling conditions: 95 °C for 3 min; 340 repeating cycles of 95 °C for 10 s (denaturation) at respective annealing temperature (Additional file 1: Table S1) for a set of primers for 30 s (annealing); 95.0 °C for 3 min, 65.0 °C for 30 s, and then a temperature increase at 0.5 °C increment to 95.0 °C (melting curve). Each assay included a no-template control and a no reverse-transcriptase control. For no template control, 1 μL of RNase/DNase free water was used as template. For the no reverse-transcriptase control, reverse-transcriptase enzyme was excluded during cDNA synthesis and resultant cDNA used as template.
A total set of 22 genes were selected for RT-qPCR analysis. Genes that are expressed in MEC during lactation and affected by the XS treatment, were selected as candidate genes for RT-qPCR analysis. A subset of DEGs obtained from RNA-seq data were also selected to validate the sequencing data. The RPL4 and RPS23 were selected as reference genes as reported in earlier study [18]. Threshold (Ct) values of target genes were normalized to the geometric mean of two reference genes to obtain ΔCt values (target gene Ct – reference gene mean Ct = ΔCt) [19]. All reactions were conducted in duplicate and Ct values were averaged for each tested gene-condition-sample combination.
For the functional annotation and pathway analyses of RNA-seq data, pathways were tested for statistical significance using a false positive threshold of 0.05 after Bonferroni’s correction. For the RT-qPCR data analysis, the raw ΔCt values of genes were log2 transformed after discovering that their values were not normally distributed (estimated by Shapiro Wilk’s Test; data not shown). Paired t-tests were employed to test significant differences among gene-condition comparisons. Data were analysed using the SPSS ver. 22 (IBM SPSS Statistics for Windows Armonk, NY). A p-value < 0.05 were considered statistically significant value.
Results
We obtained a high quality of total RNAs from goat milk fat. The total RNA concentration averaged 770.12 ± 754.7 ng/μl (mean ± SD). RNA extraction protocol provided high purity for extracted RNA having optical density (OD) ratios of 260/280 and 260/230 to 2.04 ± 0.04 and 1.84 ± 0.38 (mean ± SD), respectively. All the RNA samples of goat milk fat were suitable for RT-qPCR analysis. Samples having RNA integrity numbers (RIN) greater than 7 were sent for RNA sequencing. RIN of other RNA samples were between 6 and 7 and were suitable for RT-qPCR analysis.
Four RNA samples (RIN > 7.0) were selected for RNA sequencing from an initial group of 16 samples. The percentage of mapped reads to the ARS1 goat reference assembly [14] averaged 93.4 + 2.1 (Mean ± SD) for each sequenced sample. There were 10,267 to 11,470 genes per sample with an FPKM value ≥ 0.2, indicating some level of detectable transcription (Table 1). Functional annotation of these transcripts were enriched for cellular and metabolic pathways that were involved with catalytic and binding activities (Additional file 1: Table S2). In each of the four RNA sequencing samples, majority of the transcripts were lowly expressed (FPKM < 15; 84.3%). One animal (741) had fewer number of DEG than the other tested sample (647). This discrepancy could have been influenced by the difference in animal genetics, or due to the differences in mammary gland permeability that allowed xanthosine to diffuse into the CON gland. For abundantly expressed genes (FPKM ≥ 500), we observed an enrichment of cellular and metabolic processes (on the basis of PANTHER classification system). Transcripts corresponding to milk protein genes (CSN3, LALBA, PAEP, GLYCAM1, TPT1, PLIN2 and FABP3) were the most abundantly expressed genes. Our study showed high expression of perilipin 2 (PLIN2- a protein involved in milk fat globule formation), fatty acid binding protein (FABP3), and many ribosomal proteins during early lactation in goats. The PLIN2 gene abundantly expressed in macaque milk (Lemay et al., 2013) and FABP3 gene in cattle [20].
To characterize functions of most abundantly expressed genes (FPKM ≥500), list of genes were subjected to gene ontology (GO) classification and functional analysis using PANTHER [21]. In the TRT group, three key genes, GNB2L1, EEF1B2 and TPT1, encode key ribosome-associated proteins (Fig. 2a). Consistently, KEGG pathway analysis showed significant enrichment of ribosome-associated gene families, including the structural constituents of ribosomal genes (RPL4, RPS9, RPS10 and other ribosomal genes). Our data indicated that the highly expressed genes are mainly involved in cellular process (GO:0009987), metabolic process (GO:0008152) and cellular component organization (GO:0071840) (Fig. 2b). Additionally, milk protein genes (LALBA, CSN2, CSN3, PAEP, CSN1S1) were highly expressed in both the TRT and CON groups.
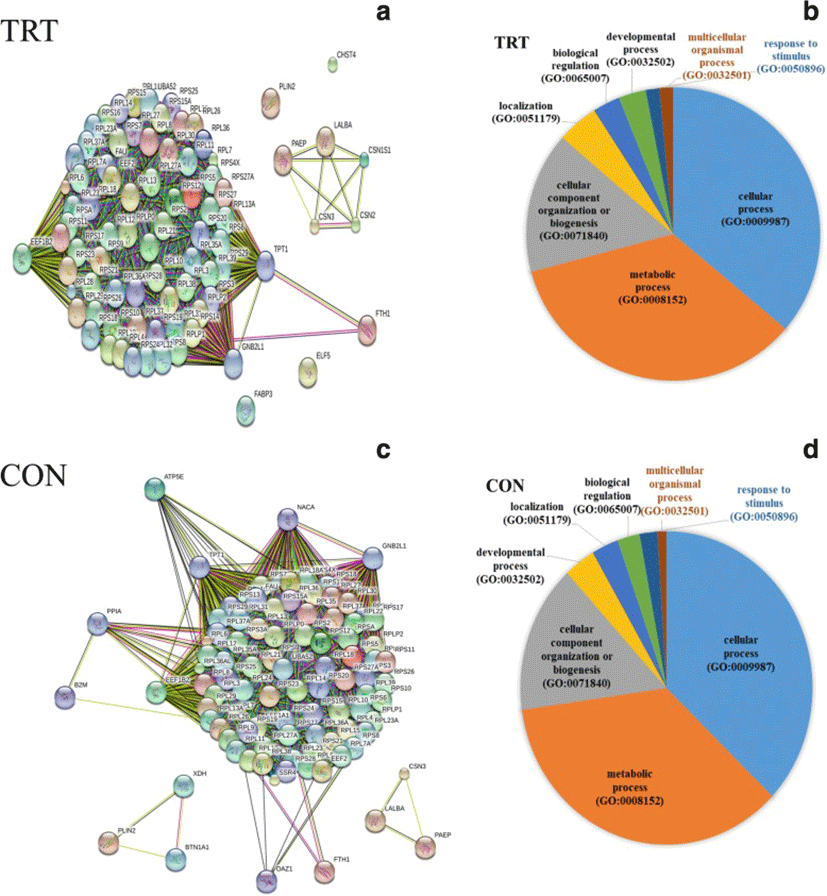
A total of 58 genes were differentially expressed MEC after XS treatment of mammary glands. A majority of DEGs (46) were found to be down-regulated after XS treatment whereas 12 genes were up-regulated (Table 2). Pathway analysis of DEG showed enrichment of genes for the following biological processes: defense response to other organsm, cytokine regulated signalling pathway, regulation of the response to other stimuli, regulation of cytokine production, and response to endogenous stimulus. The most prevalent GO-CC (cellular component) terms were associated with the cell surface (10 genes) and the plasma membrane (15 genes). Among the 10 up-regulated genes, both B2M and LTF are of particular interest. Our data did not show the up-regulation of heat shock protein genes in the TRT glands, suggesting that XS infusion did not cause mammary tissue damage.
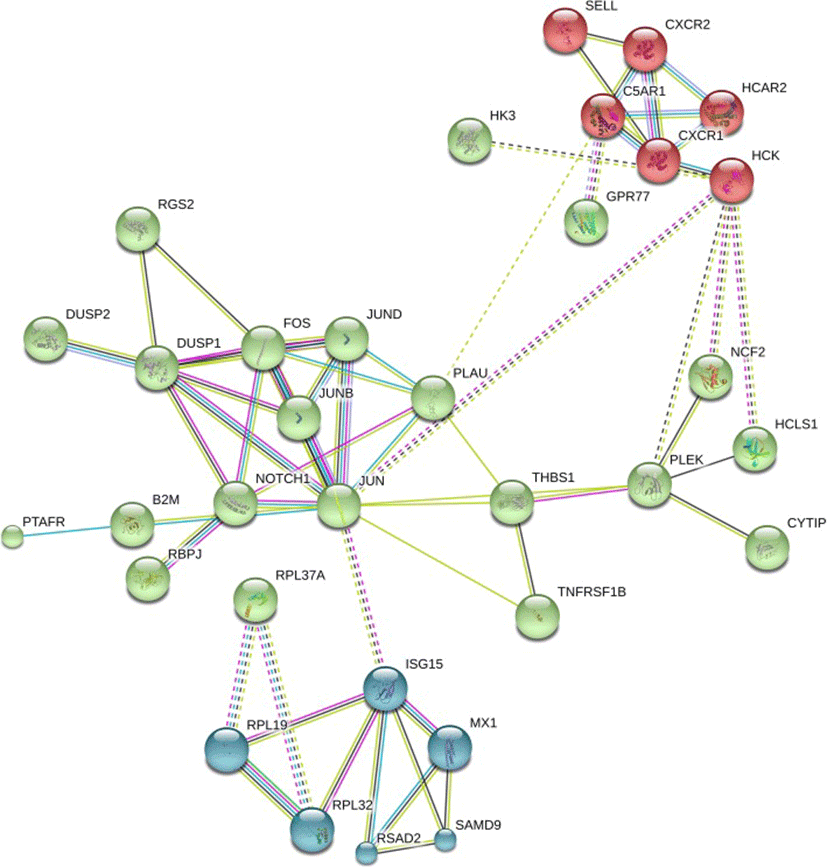
PANTHER pathway enrichment analyses showed down-regulation of inflammation signaling pathway mediated by chemokine and cytokine (CXCR1, CXCR2, PTAFR, RGS14, C5AR and JUNB) in TRT group. Notably, adhesion molecules (PECAM1, SSH2, SYNE1, CYTIP, SELL) were down-regulated upon XS treatment. Next, we investigated protein-protein interactions of DEGs using the STRING database. Out of 58 DEGs, STRING identified only 47 human homologs for calculating protein-protein interaction with a predicted confidence score of > 0.4. The interaction enrichment analysis result showed that the network has significantly more interactions (PPI enrichment p-value 2.3e-09) than the expected and the entire network contained three sub-divisions (Fig. 3) indicative of three distinct biological processes.
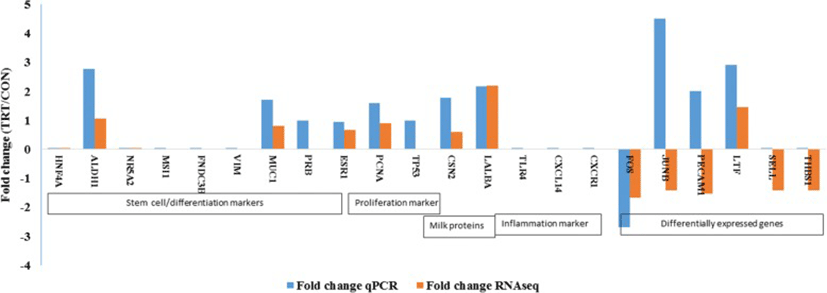
An elaborate RT-qPCR analyses of many target genes were done for three explicit purposes: 1) to validate RNAseq data; 2) to assess whether MFG RNA are quantitatively representative of the transcriptional information contained in MEC; and 3) to assess the potential effect of XS on milk production. Major milk protein genes (LALBA and CSN2), steroid receptors (ESR1 and PRB), proliferation (PCNA) and apoptosis marker (TP53) and cell differentiation marker (MUC1) were analysed in addition to DEGs.
Our RT-qPCR experiments validated many of our findings. We confirmed an absence of expression of stem cell markers (HNF4A NR5A2, MSI1, FNDC3B) that were not present in RNA-seq data. Among the list of DEGs, lactotransferrin (LTF) was up-regulated whereas, Fos proto-oncogenes (FOS) was down-regulated in TRT. RT-qPCR analyses showed similar pattern of expression as observed in DEG of RNA-seq data. However, our qPCR result was not consistent with the RNA-seq expression results of JUNB and PECAM1 genes. This difference may be attributed because of low abundance of transcripts that limits the detection by qPCR. We also failed to amplify two genes, SELL and THBS1, using RT-qPCR (Fig. 4). Later, we confirmed the extremely low abundance of SELL and THBS1 gene transcripts in milk fat RNA using droplet digital PCR (data not shown).
Discussion
The effects of XS treatment in the culture of bovine mammary epithelial cells have been shown, in vitro, to increase cell population and enhance symmetric cell division [22]. A non-invasive, in vivo method is desirable for understanding the role of XS treatment in promoting mammary epithelial cell proliferation, as an increase in the population of such cell is directly associated with milk producing ability of the glands [23]. A milk-based assay could be used to track gene expression of milk producing mammary epithelial cells [11]. It has been well documented by many researchers that the milk fat fraction is rich in RNA transcripts specific to mammary epithelial cells, as these are devoid of stromal cells, nerve cells, endotheial cells, fat cells and inflammatory cells [24]. Though the number of cytoplasmic crescents in ruminant milk is fewer in count than it is in human and macaque milk, the sensitivity of our RNA-seq analysis of milk fat isolates meant that our study was not limited by the lower proportion of cytoplasmic crescents in goat milk [25]. We demonstrate that this method provides abundant quantity and good quality of RNA suitable for RNA sequencing. This shows consistency with the findings of other studies, in which milk fat has been used to assess the transcriptome profile of milk production related genes in human [7], buffalo [26], bovine [27] and goat [28].
An advantage of our study is that XS treated (TRT) and control (CON) glands were present with the same goat. This should reduce any biological bias resulting from the sampling of different individual goats due to different genetic profiles. Thus, our experimental method captured true variation in milk producing cells affected by XS treatment, with less noise due to a different genetic background of sampled individuals. However, we cannot entirely rule out genetics as a major determinant of XS treatment response as our data also indicated that there is an individual-specific responses. This finding should be considered and evaluated for the future application of XS treatment in goats and other dairy animals for milk production.
Several genes, LALBA (α-lactalbumin), CSN2 (β-casein), CSN1S1 (α-S1-casein), CSN3 (κ-casein), GLYCAM1 (glycosylation dependent cell adhesion molecule-1) and CSN1S2 (casein-α-S2), were abundantly expressed in sheep mammary gland during lactation [29]. In our study, these genes are among the highest expressed genes found in our milk fat samples during early lactation. Additionally, we observed abundant expression of tumor protein translationally-controlled 1 (TPT1), fatty acid binding protein 3 (FABP3), perilipin 2 (PLIN2) and ribosomal proteins, which were among the top 20 abundantly expressed genes of goat mammary gland (Additional file 1: Table S3). FABP3 protein is a candidate for tumor suppressor of human breast cancer with a suggested role in arresting growth of mammary epithelial cells. TPT1 has been shown to reduce oxidative stress, minimize apotosis and promote cell survival in a p53-dependant manner [29]. PLIN2 is associated with MFG membrane and found in a wide variety of cells, including lactating mammary epithelial cells. Additionally, PLIN2 regulates lipid and protein metabolism in lactating dairy goats [30]. It is suggested that elevated expression of TPT1, PLIN2 and FABP3 might play a role in suppressing goat mammary tumor formation. Precisely, the role of FABP3 has been associated to import/export of fatty acids in the cell and highly upregulated during bovine lactation [31]. Although, reports on the incidence of mammary tumors in ruminants are rare [32], our group has encountered epithelial-mesenchymal transition, mammary epithelial cell hyperplasia and mammary pre-cancer in both goats and water buffalo [33, 34] with no sign of mammary tissue damage.
We found that XS administration twice a day for 3 days consecutively, altered the gene expression in TRT glands in goats compared to an non-inoculated CON gland on the same animal. Differential response of XS treatment was evidenced by varied number of DEGs between TRT and CON glands. The duration of XS administration (3 days) was based on an inosine study conducted in transgenic goat [35]. XS did prouduce deleterioius effects to the gland neigther causes any inflammatory reactions. We observed absence of mesenchymal cell marker (VIM), and inflammatory markers (TLR4, CXCL14 and CXCR1) amplification in the RNA harvested from mlk fat layer, indicating XS did not cause MEC inflammation. A pronounced effects of XS treatment on goat milk production may require further optimization of XS dose and duration, and may need to account for animal genetic variation. It is also imperative to note that a comparison of effects of inosine and xanthosine should be tested individually to evaluate if inosine administration is more suitable for enhancing milk production. In using Beetal goats as a test subject, the disparity of left and right mammary half gland should be considered for evaluation of true milk production potentials. The down-regulation of a dominant portion (48 out 58) of the identified DEGs, like FOS, PECAM1, JUNB, SELL, THBS1, indicated XS primarily down-regulates the expresson of genes. Platelet and endothelial cell adhesion molecule (PECAM1), selectin L (SELL) and Thrombospondin 1 (THBS1) are the cell surface adhesion molecules that mediates cell-to-cell and cell-to-matrix intereactions. Down-regulation of cell adhesion molecules is typically associated with an inhibition of cell growth [36]. Due to the limited number of goats used in this study, the number of DEG was limited. We did not find significant enrichment of KEGG pathways from the list of DEG; however, we did find several Gene Ontology (GO) terms for biological process were enriched for immune responses and cellular response to organic substance.
Previous studies have showed a pronounced effect of inosine on milk production in transgenic goats [35] suggesting that exogenous nucleosides may stimulate milk production in ruminant species. PANTHER pathway analysis revealed that several XS-induced down-regulated genes were involved in inflammation signalling pathway, which is mediated by chemokine and cytokine detection by the cells. When activated, this pathway could promote chemokine-induced adhesion and migration of leukocytes in the local tissue. We hypothesize that the down regulation of inflammatory signaling pathways induced by XS, could prevent the excessive recruitment of leukocytes to inflammatory sites. XS up-regulated expression of anti-bacterial genes (LTF and B2M), which may be beneficial to the host by preventing the incidence of mastitis. The Beta-2-microglobulin (B2M) gene codes for a protein that is associated with the major histocompatibility complex class 1 (MHC I) and is a precursor of an anti-bacterial chemokine [37]. Lactotransferrin (LTF) is a gene that encodes a major iron binding milk protein. LTF has a wide spectrum of properties that include anti-bacterial, anti-viral, and anti-cancer activities and is involved in the regulation of cellular growth [38]. Future experiments are warranted to further investigate whether XS inoculation may increase general mammary gland health in ruminants.
In our candidate gene expression analysis using RT-qPCR, we found increased expression of ALDH1A1, a mammary stem marker [39], in the TRT. We were unable to amplify two DEGs namely, SELL and THBS1 in RT-qPCR. One possibility for the inconsistency could be the low amount of transcripts in our cDNA, evidenced by Ct value of these target genes > 35 cycles. In comparison to RNA-seq, RT-qPCR has less sensitivity in accurately detecting low abundance transcripts. Droplet digital PCR (ddPCR) is useful for low abundance targets and is better than the qPCR [40]. This observation supports the facts that mammary stem cells and progenitor cells may be present in the milk [41, 42]. However, we failed to quantify the transcripts of other putative mammary stem cell markers. Absence of basal or mesenchymal cell marker gene (vimentin or VIM) expression in our study indicated that RNA extracted from milk fat is specific to luminal mammary epithelial cells but not the basal or myopethelial cells. VIM is a basal or myoepithelal cell marker. Absence of expression of inflammatory markers (TLR4, CXCR1, CXCL14) provided the evidence that RNA harvested from milk fat is mainly derived from epithelial cells but not from the polymorphonuclear cells which was consistent with previous findings [27].
Conclusions
This study characterized the global gene expression profile of goat mammary epithelial cells obtained form milk fat layer and characterized XS inoculation-associated differential gene expression in mammary epithelial cells of early lactating Beetal goat. Our study showed individual goats responded to XS treatment differently, suggesting a genetic predisposition to the treatment persists. Our findings further indicated that the administration of XS into the mammary gland has roles in down-regulation of inflammation signalling pathway, cell adhesion molecules and up-regulation of anti-bacterial genes. Down-regulation of inflammation signal and up-regulation of anti-bacterial genes may provide beneficial effects to mammary gland health. Our findings are promising. However, they were limited by a small number of goats (n = 2) included in this study, indicating that future studies with more number of animals are warranteed.