
INTRODUCTION
Oocyte aging is associated with a range of morphological, cellular and molecular changes that lead to deterioration during the aging period and negatively influence oocyte quality and subsequent embryo development in mouse [1–5], human [6,7], pig [8] and bovine [9]. Extensive studies have shown the following: The morphological changes in aged oocytes include zona pellucida hardening [10], perivitelline space enlargement [11], polar body degeneration or drifting [11] and spontaneous oocyte activation [12]. The cellular changes in aged oocytes include partial cortical granule exocytosis [13], microfilament loss [14], spindle mispairing [15] and chromosomal aneuploidy [16]. The molecular changes in aged oocytes include mitochondrial dysfunction [17,18], activity decreases of mitogen-activated protein kinase and maturation promoting factor [19], increases in reactive oxygen species [20], enhancement of apoptosis [21], alteration of DNA methylation [22] and histone modifications [23]. Together, these studies provide very important references for understanding the events that occur during oocyte aging in many species.
DNA damage includes DNA single-strand breaks (SSBs) and double-strand breaks (DSBs). The latter are generally recognized as the most important form of DNA damage [24], and should activate DNA repair and DNA damage checkpoint mechanisms [25]. DNA damage checkpoint mechanisms arrest cell division until all DNA damage is repaired [26]. However, if the damage cannot be repaired by the DNA repair mechanisms, the cell can proceed to various outcomes, including mutagenesis, cell senescence and apoptosis [27]. There are two main pathways responsible for DSB repair: homologous recombination (HR) and nonhomologous end-joining (NHEJ). In response to DNA damage, the ATM (ataxia telangiectasia mutated) and ATR (ataxia telangiectasia and rad3 related) proteins are responsible for phosphorylating histone H2A.X (H2A.X139ph) at the sites of the DNA DSBs [28,29]. The phosphorylation of histone H2A.X is closely linked to DNA DSBs repair, because the phosphorylated histone serves as a platform for the accumulation of DNA repair proteins involved in the HR (RAD51, MRE11A, BRCA1) or NHEJ (PRKDC, XRCC4, DNA ligase IV, 53BP1) pathways, which can colocalize with H2A.X at the sites of DNA DSBs [30,31].
Recently, a growing body of scientific evidence has suggested that aging is linked to the DNA damage response and DNA damage repair ability. For instance, increased DNA damage and reduced DNA damage repair in rhesus monkey granulosa cells [32] and human oocytes [33] has been associated with ovarian aging. Compared with the existing knowledge regarding somatic cells or oocytes derived from aged ovaries (reproductive or maternal aging), however, relatively little is known about the DNA damage and repair responses in postovulatory aged oocytes (postovulatory aging). A recent study that used H2A.X staining to investigate DNA damage in mice postovulatory aged oocytes found that the DNA damage level was significantly increased in mice aged oocytes [34]. However, the underlying molecular mechanisms have not been fully elucidated.
Here, we used the porcine oocyte as a model to investigate the involvement of DNA damage and repair in the function of postovulatory aging oocytes. To this end, we evaluated: 1) the levels of H2A.X and RAD51 proteins, which reflect the damage response and repair capacity, respectively; 2) the mRNA expression levels of genes involved in the HR (ATR, ATM, MRE11A, RAD51 and RAD52) and NHEJ (XRCC4, XRCC5, XRCC6, PRKDC and LIG4) pathways of DNA repair; 3) the mRNA abundance of genes involved in cell cycle control (CHK1 and CHK2); 4) the level of H3K79me2 in aged oocytes; and 5) the embryonic developmental capacity of porcine aged oocytes.
MATERIALS AND METHODS
All utilized chemicals and reagents were obtained from Sigma (St. Louis, MO, USA) unless otherwise stated. All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Chung Nam National University, Korea (202003A-CNU-002).
Porcine ovaries were obtained from a local abattoir and transported to the laboratory in physiological saline at 35°C within 2 h of collection. Follicular contents were aspirated from antral follicles (3 to 6 mm in diameter) visible on the ovarian surface using an 18-gauge needle attached to a 10-ml disposable syringe. Cumulus oocyte complexes (COCs) were collected and washed two or three times in phosphate-buffered saline (PBS) (Gibco, USA) containing 0.1% polyvinyl alcohol (PVA). Only oocytes with a uniform ooplasm and compact cumulus cell mass were used for in vitro maturation.
For IVM, the COCs were washed three times and groups of about 50~60 COCs were matured in 500 μL maturation medium (tissue culture medium [TCM] 199 containing 10% porcine follicular fluid, 3.5 mM D-glucose, 0.57 mM L-cysteine, 0.91 mM sodium pyruvate, 75 μg/mL penicillin, 50 μg/mL streptomycin, 10 ng/mL epidermal growth factor, 10 IU/mL pregnant mare serum gonadotropin, 10 IU/mL human chorionic gonadotropin) in each well of a four-well multi dish in saturated-humidity air containing 5% CO2 at 38.5°C for 44 h. For IVA, oocyte aging was performed as described previously [35]. In brief, the matured porcine oocytes were cultured in the same medium and condition for an extended period of 24 h (IVA 24 h) or 48 h (IVA 48 h) to mimic postovulatory oocyte aging. After oocyte IVM or IVA, we removed cumulus cells from COCs by pipetting or vortexing them in 0.1% hyaluronidase solution. Survival of oocytes was investigated under a stereomicroscope based on oocyte morphology. Only oocytes with integrated oolemma and zona pellucida and a polar body were used for experiments, whereas those with lysis of the oolemma or damage to the zona pellucida or cytoplasmic fragmentation were considered to discard.
For PA, the oocytes were subjected to electrical activation. Cumulus cell-free oocytes were washed three times and then equilibrated in an activation solution containing 0.3 M D-mannitol, 0.1 mM MgSO4, 0.05 mM CaCl2 and 0.01% PVA. The oocytes were placed between the platinum electrodes in activation solution, and activation was induced with two direct current pulses of 1.1 kV/cm for 30 μs, using an Electro Cell Manipulator 2001 (BTX, San Diego, CA, USA). After electric stimulation, the oocytes were cultured in porcine zygote medium-3 containing 3 mg/mL bovine serum albumin (BSA) and 7.5 μg/mL cytochalasin B (CB) for 5 h (to inhibit extrusion of the second polar body) at 38.5°C in a humid 5% CO2 atmosphere. After 5 h incubation with CB, the embryos were transferred to CB-free culture medium for further culture.
Immunofluorescence staining was performed as described previously [36]. Briefly, oocytes were washed in PBS-PVA and then fixed in 4% paraformaldehyde for 30 min. After being permeabilized with 0.5% (v/v) Triton X 100 in PBS-PVA for 30 min, they were blocked with 3% BSA for 1 h. Samples were washed in PBS containing 0.5% BSA and 0.1% gelatin (PBG), and incubated overnight at 4°C with primary antibodies. The samples were washed in PBG and then reacted with secondary antibodies in the dark for 1 h. After being washed with PBG, the samples were mounted on glass slides using Vectashield mounting medium with 4’,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA, USA) and viewed under a Zeiss laser-scanning confocal microscope (LSM5 Live, Carl Zeiss, Oberkochen, Baden-Württemberg, Germany). Negative control embryos were processed as described above, except that no primary antibody was added. The utilized primary antibodies recognized H2A.X (Merck KGaA, Darmstadt, Germany), RAD51 (Santa Cruz Biotechnology, Dallas, TX, USA), H3K79me2 (Abcam, Cambridge, UK), and H3 (Active Motif, Carlsbad, CA, USA). The utilized secondary antibodies were goat anti-mouse IgG-R (Santa Cruz Biotechnology), goat anti-mouse IgG-FITC (Santa Cruz Biotechnology), or donkey anti-rabbit FITC (Abcam).
EdU staining was performed using a Click-iT EdU Imaging kit (Invitrogen, Eugene, OR, USA). The provided manufacturer’s instructions are normally intended for use in cell culture, but the protocol was adapted for porcine embryos as follows. Briefly, porcine blastocysts were collected and incubated in EdU (10 μM) solution for 3 h, and then fixed in 4% paraformaldehyde for 15 min. After being washed with 3% BSA, samples were permeabilized with 0.5% Triton X-100 for 20 min. The samples were again washed with 3% BSA and then reacted with a Click-iT reaction cocktail containing Click-iT reaction buffer, CuSO4, reaction buffer additive and Alexa Fluor 488 azide for 30 min in the dark. After EdU detection, the samples were washed and mounted on glass slides using VECTASHIELD mounting medium containing DAPI. Images were captured using a Zeiss laser-scanning confocal microscope.
Total RNA was extracted from each sample (200 oocytes) using an RNeasy Mini Kit (Qiagen, Valencia, CA, USA) and an RNase-Free DNase Set (Qiagen) in accordance with the manufacturer’s instructions. cDNA was synthesized from the isolated total RNA using a TOPscript™ RT DryMIX kit (Enzynomics, Daejeon, Korea). Quantitative real-time PCR was performed using a TOPreal™ qPCR 2X PreMIX (SYBR Green with low ROX) kit (Enzynomics) on a CFX96 Touch Real-Time PCR Detection System (Bio-Rad, St. Ingbert, Germany). PCR controls run with no template were performed for each primer pair. Finally, the relative mRNA expression levels of each gene were analyzed using the 2-△△Ct method [37]. The primer sequences used for real-time PCR are presented in Table 1.
DNA damage was detected by detecting DNA double strands breaks using an antibody against H2A.X. Four independent experiments were performed, and total 40 (Control), 42 (IVA 24 h), 46 (IVA 48 h) oocytes were analyzed. DNA repair was evaluated using an antibody against RAD51. Three independent experiments were performed, and total 30 (Control), 30 (IVA 24 h), 30 (IVA 48 h) oocytes were analyzed in this set of experiment. Fluorescence intensities were analyzed, and the background value was subtracted using the ImageJ software.
Expression levels of genes involved in the HR (ATR, ATM, MRE11A, RAD51 and RAD52) and NHEJ (XRCC4, XRCC5, XRCC6, PRKDC and LIG4) pathways for DNA damage repair and cell cycle checkpoint (CHEK1 and CHEK2) in porcine aged oocytes were investigated using real-time quantitative PCR. Three independent experiments were performed in this set of experiment. Each experimental group contains about 200 oocytes were used for total RNA extraction.
The expression of H3K79me2 was detected using antibody against H3K79me2. Four independent experiments were performed, and total 48 (Control), 52 (IVA 24 h), 52 (IVA 48 h) oocytes were analyzed. The expression of H3 was detected using antibody against H3. Three independent experiments and total 30 (Control), 30 (IVA 24 h), 30 (IVA 48 h) oocytes were analyzed. Immunofluorescence intensities were analyzed, and the background value was subtracted using the ImageJ software.
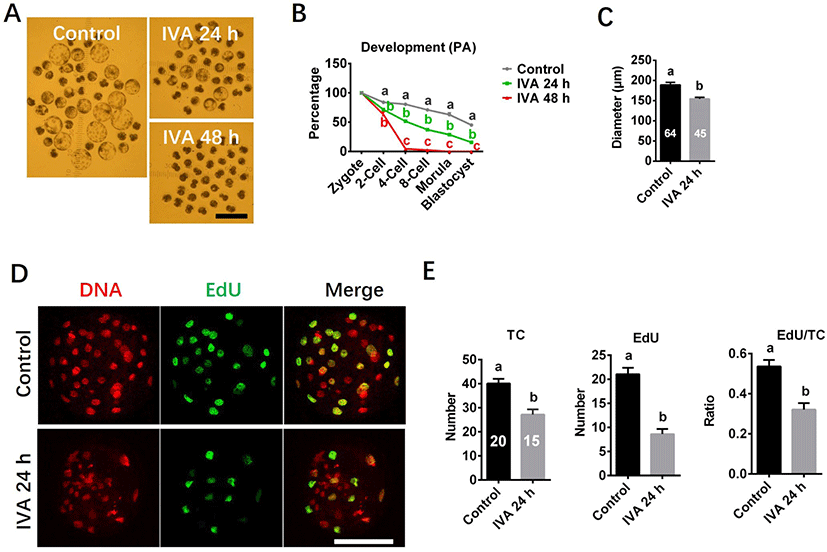
Developmental capacity and cell proliferation potential were investigated using morphology and EdU staining. Blastocyst morphology pictures were captured under a stereomicroscope with an ocular scale at day 7 of culture, and diameter of blastocyst was measured using the ImageJ software. At least three independent experiments were performed for each study. Total 189 (Control), 180 (IVA 24 h), 193 (IVA 48 h) oocytes were analyzed for embryo development. Total 64 (Control), 45 (IVA 24 h) blastocysts were analyzed for blastocyst diameter. Total 20 (Control), 15 (IVA 24 h) blastocysts were analyzed for cell proliferation potential in blastocyst. Because IVA 48 h group failed to develop to blastocyst stage, the analyses presented in both diameter and cell proliferation potential of blastocyst were not performed for the IVA 48 h group.
Statistical analyses were conducted using SPSS 17.0 (SPSS, Chicago, IL, USA). At least three replicates were performed for each experiment. Percentage data were subjected to arcsine transformation prior to analysis. All experimental data were compared by one-way ANOVA followed by Fisher’s protected least significant difference test or Student’s t-test. The data are expressed as the mean ± sem. p < 0.05 was considered significantly different.
RESULTS
We first assessed the DNA damage response protein, H2A.X139ph, to evaluate the DNA DSB response in porcine aged oocytes. The fluorescence signals of H2A.X139ph (green) could be detected in all porcine oocytes (Fig. 1A); however, a higher level of DNA damage response was observed in porcine aged oocytes (IVA 24 h and IVA 48 h) than in control oocytes (Fig. 1B). To examine the activity of the DNA damage repair response during the oocyte aging period, we immunostained for RAD51 in porcine aged oocytes. The fluorescence signals of RAD51 (red) could be detected in porcine oocytes (Fig. 1C), and the relative intensities of fluorescence signals were significantly lower in aged oocytes than in controls (Fig. 1D).
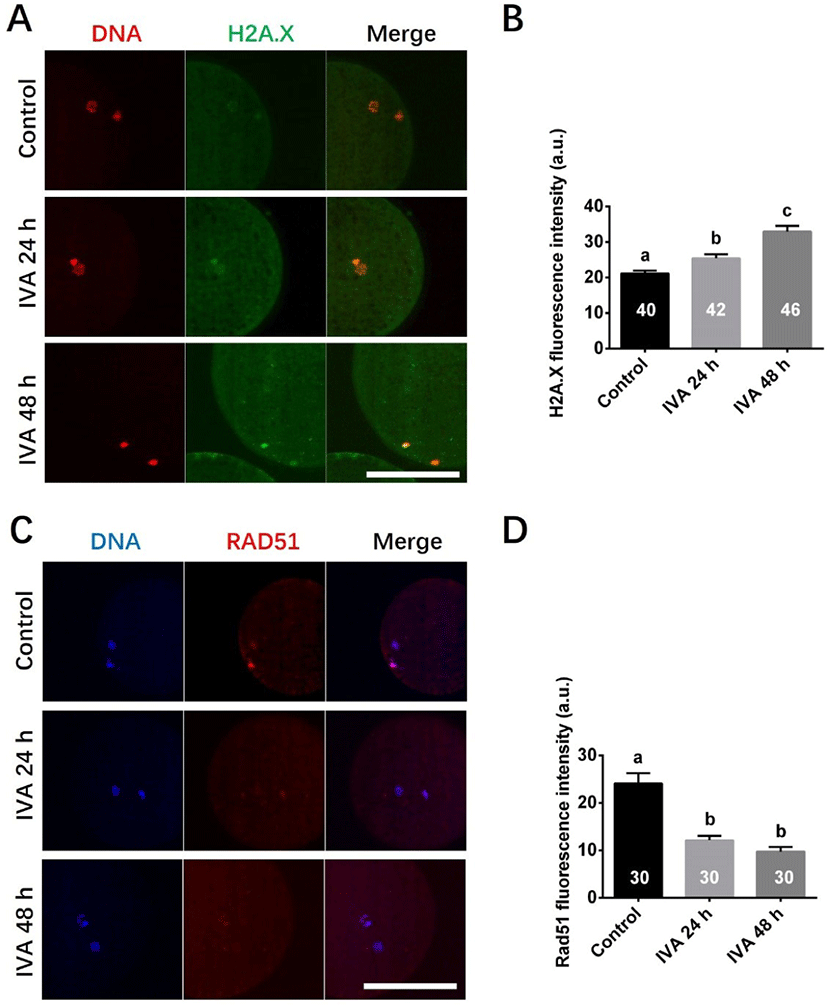
To test whether the increased DNA damage in aged oocytes could be related to a deficiency of DNA damage repair, we measured the mRNA abundances of genes involved in the HR (ATR, ATM, MRE11A, RAD51 and RAD52) and NHEJ (XRCC4, XRCC5, PRKDC, LIG4 and XRCC6) pathways of DNA damage repair in porcine aged oocytes. The levels of various genes involved in the HR repair pathway were found to be significantly decreased in the IVA 24 h group (ATR, MRE11A and RAD52) and/or IVA 48 h group (ATR, ATM, MRE11A, RAD51 and RAD52) when compared to controls (Fig. 2A). The expression levels of various genes involved in the NHEJ pathway were also significant lower in the IVA 24 h group (XRCC4, PRKDC and LIG4) or IVA 48 h group (XRCC4, XRCC5, PRKDC and LIG4) than in controls (Fig. 2B). However, there was no difference in the level of the XRCC6 gene between the control and aged oocyte groups (Fig. 2B).
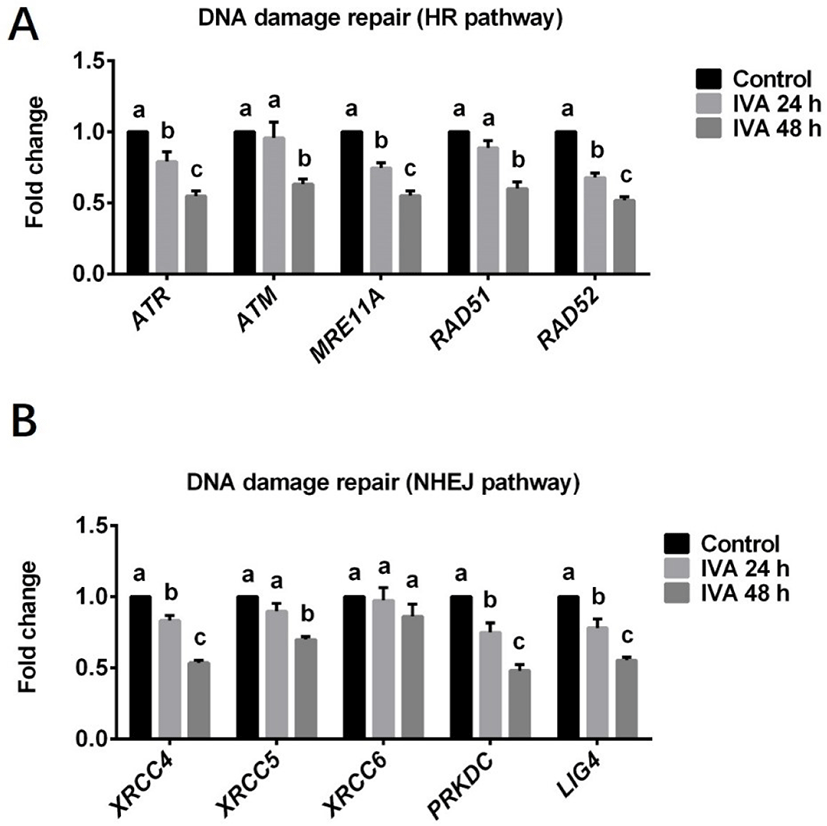
The transcript levels of the cell cycle checkpoint genes CHEK1 and CHEK2 were also estimated in porcine aged oocytes (Fig. 3). The mRNA level of CHEK1 was significantly higher in aged oocytes (both IVA 24 and 48 h) than in controls (Fig. 3A). The expression level of CHEK2 did not significantly differ between the IVA 24 h group and the control group, but it was significantly increased in the IVA 48 h group compared to the control group (Fig. 3B).
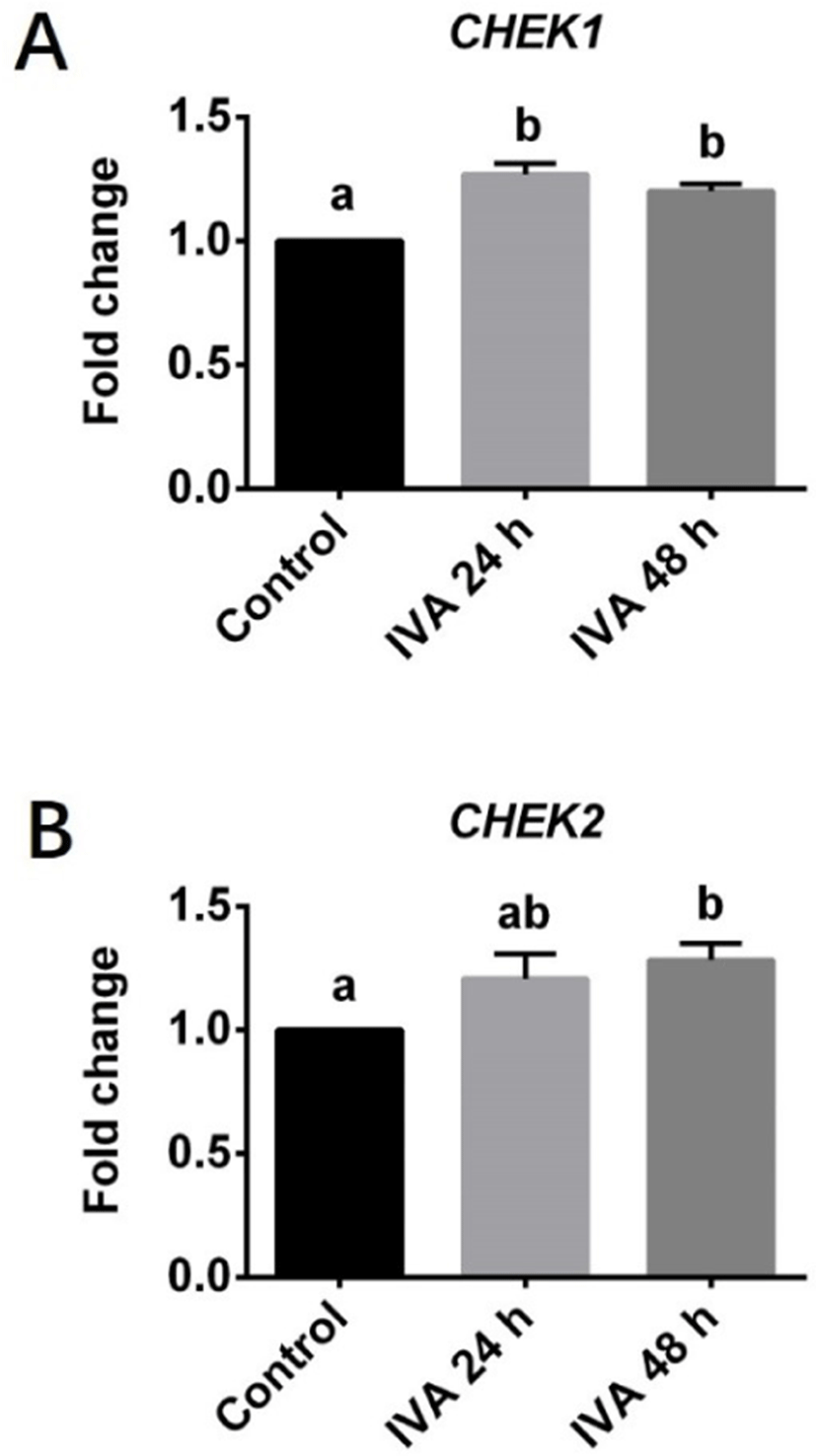
The expression levels of H3K79me2 and H3 were examined in porcine aged oocytes, as shown in Fig. 4. Immunofluorescence revealed that the expression level of H3K79me2 was significantly decreased in porcine aged oocytes (IVA 24 h and IVA 48 h) when compared to controls (Figs. 4A and B). In contrast, H3 exhibited similar expression levels in the control, IVA 24 h and IVA 48 h groups (Figs. 4C and D).
The embryo development capacity was significantly reduced in the aged oocyte groups compared to controls (Fig. 5). Embryos derived from the IVA 24 h group could develop to the blastocyst stage, but the rate of blastocyst formation was significantly lower than in the control group (Figs. 5A and B). Blastocysts derived from the IVA 24 h group also had smaller diameters than those derived from control oocytes (Fig. 5C). In contrast, no blastocyst formation was observed among the IVA 48 h group, and more than half of the derived embryos arrested at the 2-cell stage by day 7 of culture (Figs. 5A and B). EdU staining showed that the total cell number (TC), the EdU-positive (S-phase) cell number and the ratio of EdU-positive cells to TC were significantly lower in blastocysts derived from the IVA 24 h group compared to the control values (Figs. 5D and E).
DISCUSSION
Oocyte aging is known to negatively influence oocyte quality, embryonic development and reproductive outcomes. However, the underlying molecular mechanisms have not been fully elucidated. In the present study, we investigated whether the DNA damage response and DNA damage repair ability are closely linked to oocyte aging. Our results showed that postovulatory oocyte aging increased the DNA damage response and suppressed the DNA damage repair capacity in porcine oocytes. The development of therapies that target these signaling pathways might help prevent or delay oocyte aging.
Previous reports suggested that an increased DNA damage response and reduced DNA repair capacity in rhesus monkey granulosa cells [32] or mouse and human oocytes [33] may contribute to ovarian aging. In mouse aged oocytes derived from an in vitro aging model, researchers observed a considerable increase in the level of DNA damage in comparison with control oocytes [34]. However, the previous studies had notable limitations, such as the use of oocytes or cells from aged ovaries or the use of H2A.X staining alone to evaluate the DNA damage response. Here, in a pig in vitro oocyte aging model, we first assessed the DNA damage response marker, H2A.X, and the repair protein, RAD51, in porcine aged oocytes. The presence of phosphorylated histone H2A.X (H2A.X 139ph) foci has been widely used to estimate the occurrence of DNA damage in somatic cells [38,39], oocytes [40] and embryos [25,31]. RAD51, a recA homolog that binds the single-strand DNA generated by the Mre11-Rad50-NBS1 complex, is a key factor for DNA damage repair [41]. In the present study, porcine oocytes showed an appreciable H2A.X signal (consistent with an active DNA damage response) that increased in intensity as the oocytes aged, indicating that the incidence of DNA damage in porcine oocytes increased with the aging time. Conversely, the protein expression of RAD51, which reflects the damage repair ability, decreased in aged oocytes over time. Our results suggest that oocyte aging increases DNA damage and suppresses the DNA damage repair ability in pig.
To further evaluate the influence of oocyte aging on the DNA damage repair ability, we investigated the mRNA expression levels of genes involved in the NHEJ (XRCC4, XRCC5, XRCC6, PRKDC and LIG4) and HR (ATR, ATM, MRE11A, RAD51 and RAD52) pathways in porcine aged oocytes. Our results showed that the relative mRNAs abundance of the genes (except XRCC6) involved in the two repair pathways decreased over time in porcine aged oocytes, suggesting that these two DNA damage repair pathways are suppressed in this model. However, there was no difference in the level of the XRCC6 gene between control and aged oocytes, and it is speculated that porcine oocyte aging in vitro suppresses DNA damage repair ability probably not by XRCC6 gene product. Consistent with our results, previous studies that examined the relationship between DNA damage repair and ovarian aging in human oocytes [24] and rat primordial follicles [42] found that the expression levels of some important DNA repair genes (e.g., BRCA1, RAD51 and MRE11) declined with age. Together, the previous and present results suggest that it may be difficult for the DNA damage repair mechanisms to repair aging-induced DNA damage. Supplementation of antiaging chemicals Coenzyme Q10 and melatonin could reduce DNA damage in mice [34] and bovine [9] aged oocytes by detecting DNA double strands breaks using an antibody against H2A.X had been already reported. However, there is little evidence that adding antiaging chemicals improves DNA damage repair ability. Future work is needed to examine whether the DNA repair ability could be improved by using antiaging treatment in oocytes.
Cell cycle checkpoint kinase 1 (Chk1) and 2 (Chk2) are well known to be activated by the ATM and ATR kinases in response to DNA damage; this leads to cell cycle arrest, which allows the cell sufficient time to repair the damaged DNA before it undergoes replication and segregation in cleaving cells [25,43,44]. Once the DNA damage repair is complete, the cell cycle checkpoint proteins are inactivated and the cell cycle resumes. In the current study, we found that the mRNA expression levels of CHEK1 and CHEK2, which encode Chk1 and Chk2, respectively, were increased along with the DNA damage response in porcine aged oocytes compared to controls. This is in line with previous observations that CHEK1 and CHEK2 are activated in mouse [45] and pig [25] embryos that have increased levels of DNA damage. The CHEK1 and CHEK2 genes are usually activated to enable the damaged DNA to be repaired. However, we found that the expression levels of DNA damage repair-related proteins and genes were decreased in aged oocytes. We speculate that the long-term arrest of aged oocytes at metaphase II stage may allow DNA damage accumulate to a level at which the DNA damage repair ability becomes suppressed.
Epigenetics has recently emerged as another possible determinant of (and potentially a major contributor to) the aging phenotype [46]. Generally, changes in histone methylation are related to the activation/repression of gene transcription [47]. H3K79me2, which has been suggested as a marker of active genes in mammalian cells [48], and is also thought to be associated with the DNA damage repair mechanism [49]. Here, we found that the expression level of H3K79me2 in porcine oocytes was obviously decreased during in vitro postovulatory aging. We speculate that this may be associated with the observed deficiency in the DNA damage repair mechanism of porcine aged oocytes.
Previous studies found that laser microbeam-induced DNA damage in mouse embryos reduced the rates of cleavage and blastocyst formation [45], and the incidence of the DNA damage response is higher and developmental capacity is lower in late-cleaving porcine embryos than in their early-cleaving counterparts [25]. The above studies imply that there is a negative relationship between DNA damage and embryo development. In the present study, we found that embryos derived from aged oocytes have decreased developmental abilities: the IVA 24 h group showed a decreased ability to develop to the blastocyst stage, while the IVA 48 h group failed to develop to the blastocyst stage, with more than half of the embryos arresting at the 2 cell stage. Thus, we speculated that the presence of DNA damage induced by oocyte aging could be one of the reasons causing embryo development impaired. Moreover, consistent with previous reports that the kinetics of cleaving [25] and blastocyst formation [50] can influence the total cell number in blastocysts, we found that blastocysts derived from aged oocytes possess lower total cell and EdU-positive cell numbers than controls. Thus, we suggest that oocyte aging suppresses the cell proliferation capacity in porcine blastocysts.
In conclusion, we herein demonstrate that oocyte aging increased the DNA damage response in porcine aged oocytes as indicated by upregulation of H2A.X expression. However, the DNA damage repair ability was suppressed in porcine aged oocytes matured in vitro, downregulation of the RAD51 protein level and the mRNA expression levels of genes involved in both HR and NHEJ pathways. The expression levels of the cell cycle checkpoint genes, CHKE1 and CHKE2, were upregulated in porcine aged oocytes in response to induced DNA damage. The H3K79me2 level decreased in porcine oocytes during in vitro postovulatory aging. In addition, postovulatory oocyte aging altered the kinetics of both cleavage and blastocyst formation and suppressed the cell proliferation capacity in blastocysts. These results provide useful information to help us understand the internal events that govern oocyte aging and thus may be targeted to delay oocyte aging.