
INTRODUCTION
The weaning process represents one of the most significant stressors in swine production, particularly affecting piglets at 3–4 weeks of age, when their gastrointestinal and immune systems are still immature. During this critical transition period, weaned piglets undergo substantial physiological changes including disruptions in gut integrity, alterations in villus architecture and mucosal permeability, and shifts in gut microbiota composition [1]. These changes primarily driven by dietary transitions increase the susceptibility of piglets to infectious diseases [2,3]. Escherichia coli is considered one of the major enteric pathogens infecting weaned piglets, and infection can lead to diseases such as post-weaning diarrhea (PWD). PWD is primarily caused by F4 or F18 adhesin-type E. coli strains. Infected pigs typically show severe diarrhea resulting in increased mortality, growth retardation, and economic losses [4,5].
Phytobiotic supplementation has been explored as a potential strategy to prevent or mitigate diseases caused by pathogenic bacteria during the weaning period. Phytobiotics are natural bioactive compounds derived from various plants that support animal health, promote overall growth, and provide protection against infectious diseases [6]. To date, more than 5,000 phytobiotics have been identified from diverse sources such as herbs, essential oils, and agricultural by products. Phytobiotics can be administered in various forms, including dried materials, powders, extracts, or solid formulations. Phytobiotics are generally classified into four categories based on their origin and processing characteristics: 1. Herbs (flowering, non-woody, and non-perennial plants); 2. Spices (plants with strong aromas or flavors); 3. Essential oils (volatile lipophilic compounds); 4. Oleoresins (extracts derived from non-aqueous solutions).
The efficacy of phytobiotics as feed additives (PFAs) for pigs has been extensively studied. Numerous reports have demonstrated that dietary inclusion of PFAs improved growth performance in pigs, which was largely attributed to enhanced nutrient digestibility and improved intestinal morphology [7,8]. In addition to their direct effects on intestinal tissues, phytobiotics can modulate gut microbiota composition, with different compounds exerting distinct effects. For example, carvacrol, a phenolic compound found in black pepper and thyme, showed antimicrobial activity comparable to that of conventional antibiotics by reducing bacterial load and suppressing microbial activity in the gastrointestinal tract [9]. Its mechanisms of action include disruption of bacterial cell wall integrity, inhibition of nucleic acid and protein synthesis, and compromise of membrane permeability [10,11]. These antimicrobial effects have been associated with reductions in Salmonella and E. coli counts in pig feces, along with decreased incidence of diarrhea [12,13]. Conversely, certain phytobiotics promote beneficial microbial populations. Essential oil blends derived from oregano, anise, and citrus peel have been shown to increase the abundance of lactic acid bacteria, thereby enhancing intestinal fermentation capacity [14]. Similarly, supplementation with coix seed has been reported to significantly increased the abundance of Lactobacillus and Bacteroides in the gastrointestinal tract of weaning piglets. This effect is likely due to its rich composition of starch, oil, polysaccharides, and proteins [15].
Collectively, these findings suggest that phytobiotics not only enhance intestinal integrity and function but also serve as fermentation substrates for beneficial gut microbiota, thereby contributing to host health. As a result, there is growing interest in their application as alternatives to antibiotics, particularly during the weaning period, a time when immune competence is still developing and pigs are highly vulnerable to enteric infections. Despite their potential, comprehensive studies examining the specific effects of phytobiotics on gut health and microbial composition remain limited. Therefore, the present study aimed to investigate the effects of dietary supplementation with phytobiotic compounds on intestinal immunity and gut microbiota composition in weaned piglets challenged with pathogenic E. coli.
MATERIALS AND METHODS
Five phytobiotic materials, labeled P1 through P5, were used in both in vitro and in vivo experiments. All materials were procured from Eugene-Bio. The compositions of the phytobiotic treatments were as follows: P1: bitter citrus extract containing 25%–27% naringin and 11%–15% neohesperidin (BioFlavex GC, HTBA); P2: microencapsulated blend containing 7% thymol and 7% carvacrol (Avipower 2, VetAgro SpA, Reggio Children); P3: mixture containing 40% P1 + 10% P2 + 50% excipient; P4: premixture of grape seed, grape marc extract, green tea, and hops containing 10% flavonoids (AntaOx Flavosyn, DR. Eckel GmbH); P5: fenugreek seed powder containing 12% saponin (Fenugreek Seed Powder, P&D Export).
The Raw 264.7 murine macrophage cell line (Cat. No AC28116) was obtained from the Korean Collection for Type Cultures (KCTC). Cells were seeded at a density of 1 × 10⁵ cells/well in 500 μL of culture medium in 24-well plates (Corning). Dulbecco’s Modified Eagle’s Medium (DMEM; Gibco) supplemented with 10% fetal bovine serum (FBS; Gibco) and 1% antibiotics (100 U/mL penicillin and 100 µg/mL streptomycin) was used for cultivation. Cells were incubated at 37°C in a humidified atmosphere containing 5% CO2 for 24 hours. Following the initial incubation, 100 μL of each phytobiotic was added to the wells, and cells were then incubated for an additional 24 hours under the same conditions. Triton X-100 was used as a positive control.
To measure the expression levels of TNF-α, NF-κB (p50), and NF-κB (p65), total RNA was extracted using the NucleoSpin® RNA kit (MACHEREY-NAGEL) after washing the cells twice with 500 μL of 1× DPBS. RNA concentration and purity were measured using a Colibri Microvolume Spectrometer (Titertek Berthold). Subsequently, the RNA was reverse transcribed into complementary DNA (cDNA) using the AccuPower® RT PreMix (Bioneer) according to the manufacturer’s protocol.
Quantitative real-time polymerase chain reaction (qRT-PCR) was conducted using the CFX Connect™ Real-Time System (Bio-Rad) to quantify gene expression levels. The qRT-PCR cycling conditions were as follows: initial denaturation at 95°C for 30 seconds, followed by 40 cycles of denaturation at 95°C for 10 seconds and annealing at 60°C for 10 seconds. The reaction concluded with a final step at 65°C for 5 seconds, followed by 95°C. Expression levels of genes associated with immune and inflammatory responses were normalized to the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and relative expressions were compared across experimental groups. Primer sequences used in this study are listed in Table 1.
The animal experiment was approved by the Institutional Animal Care and Use Committee of Chungbuk National University, Cheongju, Korea (Approval No. CBNUA-1618-21-02). A total of 63 weaned piglets ([Yorkshire × Landrace] × Duroc), 28 days of age with an initial average body weight (BW) of 8.03 ± 0.43 kg, were used in a three-week (21-day) experiment. Piglets were randomly allocated to seven treatment groups based on their initial body weight and E. coli challenge status. Each treatment group consisted of nine replicate cages, with one castrated piglet housed per cage. All piglets were housed in individual stainless steel metabolic cages (45 × 55 × 45 cm) under optimized environmental conditions. The experimental treatment groups were as follows: NC, negative control (basal diets without E. coli challenge); PC, positive control (basal diets + E. coli challenge); T1, PC + 0.04 % P1; T2, PC + 0.01 % P2; T3, PC + 0.10 % P3; T4, PC + 0.04 % P4; T5, PC + 0.10 % P5. The basal diet was formulated to meet the nutritional requirements of weaned piglets. The ingredient composition and nutrient contents of the diets are provided in Table 2.
Throughout the 21-day experimental period, piglets had ad libitum access to water, and feed was provided twice daily at 08:30 and 17:30. The feed was mixed with water in a 1:1 ratio immediately before feeding. The E. coli challenge was administered orally from days 8 to 10 by delivering 10 mL of nutrient broth containing E. coli at a concentration of 1.2 × 10¹⁰ CFU/mL.
At the end of the experiment (day 21), piglets were euthanized using carbon dioxide gas followed by exsanguination. Intestinal tissue samples were collected from the ileum and colon, at least 10 cm distal to the cecum. The collected samples were rinsed with phosphate-buffered saline (PBS), and mucosal tissues were carefully scraped using sterile scalpel blades. All samples were immediately stored at −80°C for subsequent analysis.
Total RNA was extracted from the mucosal samples using the NucleoSpin® RNA kit (MACHEREY-NAGEL) according to the manufacturer’s instructions. RNA concentration and purity were assessed using a Colibri Microvolume Spectrometer (Titertek Berthold). cDNA was synthesized from the extracted RNA using the AccuPower® RT PreMix (Bioneer) following the manufacturer’s protocol.
qRT-PCR was performed using the CFX Connect™ Real-Time System (Bio-Rad) under the following thermal cycling conditions: initial denaturation at 95°C for 30 seconds, followed by 40 cycles of denaturation at 95°C for 10 seconds and annealing at 55°C for 10 seconds. The reaction concluded with an extension step at 55°C for 5 seconds and a final extension at 95°C.
Gene expression levels of the tight junction protein ZO-1 and mucins (MUC1, MUC2, and MUC3) in the ileal and colonic mucosa were analyzed. Primer sequences used were listed in Table 3. Expression levels were normalized to the housekeeping gene GAPDH and compared across treatment groups.
Fecal samples were collected from three randomly selected piglets per treatment group before the E. coli challenge (day 8) and at the end of the experiment (day 21). Fecal samples were collected directly from the rectum of each pig using sterile gloves resulting in a total of 42 samples from 21 piglets. All fecal samples were immediately stored at −80°C until further analysis.
For microbial community analysis, total DNA was extracted from 200 mg of feces using the QIAamp Fast DNA Stool Mini Kit (QIAGEN) according to the manufacturer’s instructions. The concentration and purity of the extracted DNA were measured using a Colibri Microvolume Spectrometer (Titertek Berthold). DNA purity with an optical density (OD) ratio of 260/280 between 1.8 and 2.0 was considered to be of high purity and acceptable for downstream applications.
For amplicon sequencing of the V5–V6 hypervariable regions of the 16S rRNA gene, PCR was performed using primers 799F-mod6 (5’-CMGGATTAGATACCCKGT-3’) and 1114R (5’-GGTTGCCTCGTTGC-3’) [16]. Each 50 μL PCR reaction contained KOD One™ PCR Master Mix -Blue- (TOYOBO), 10 pmol of each primer, and 5 ng/μL of template DNA. The PCR cycling conditions were as follows: initial denaturation at 98°C for 3 minutes, followed by 25 cycles of denaturation at 98°C for 10 seconds, annealing at 57°C for 5 seconds, and extension at 68°C for 1 second, with a final extension at 72°C for 5 minutes. The amplified PCR products were purified using the Wizard® SV Gel and PCR Clean-Up System kit (Promega). Barcoded 16S rRNA gene amplicons were then sequenced on the Illumina MiSeq platform by BRD Korea.
Raw 16S rRNA gene sequencing data were analyzed using the Quantitative Insights into Microbial Ecology 2 (QIIME2) software package [17]. Quality filtering was performed based on a PHRED quality score threshold of 27 to remove low-quality reads and sequences with ambiguous base calls, thereby minimizing the influence of random sequencing errors. The deblur plugin was used to trim sequences to a uniform length of 300 bp, after which amplicon sequence variants (ASVs) were inferred to represent true biological sequences.
For phylogenetic diversity analysis, multiple sequence alignment was conducted using the multiple alignment using fast Fourier transform (MAFFT) pipeline. Alpha diversity metrics were calculated to evaluate species richness and evenness within individual samples. Beta diversity was analyzed to compare microbial community composition between groups, using both weighted (quantitative) and unweighted (qualitative) UniFrac distance metrics. Differences in community structure were visualized through principal coordinate analysis (PCoA) plots. Taxonomic classification of ASVs was performed using a naïve Bayesian classifier trained on the Ribosomal Database Project (RDP) reference database, version 19.
Statistical analyses were performed using GraphPad Prism version 8.00 for Windows (GraphPad Software). Significant differences in experimental parameters among treatment groups were assessed using the Kruskal–Wallis H test. Dunn’s multiple comparison test was used to determine pairwise differences between treatment groups. To evaluate differences in microbial community structure among experimental groups, the analysis of similarities (ANOSIM) method was applied.
RESULTS
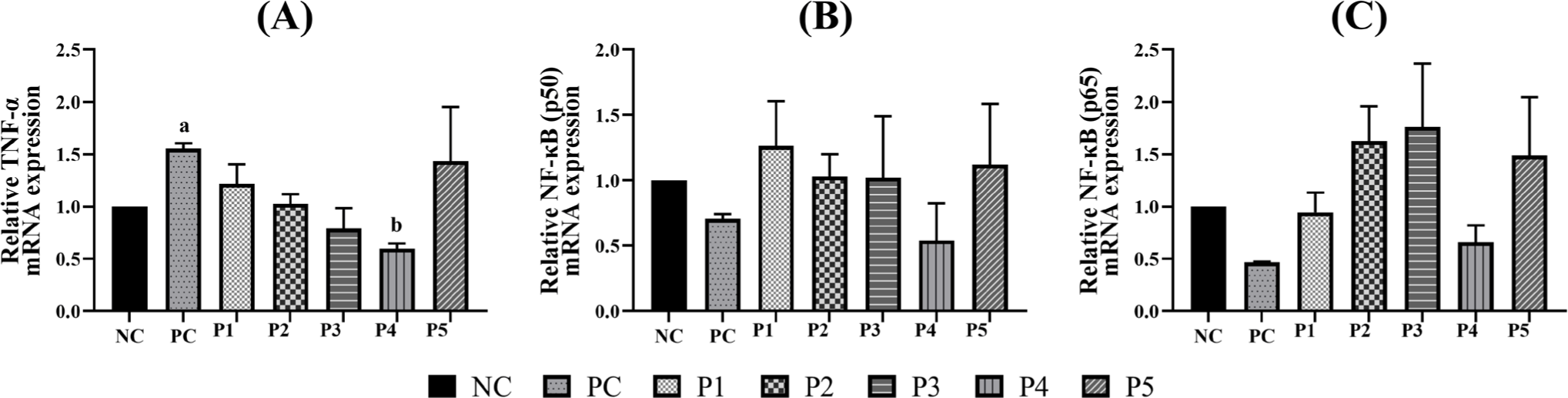
The effects of phytobiotics on the expression of immune and inflammation-related markers including TNF-α and NF-κB were evaluated in RAW 264.7 cells (Fig. 1). Treatment with Triton X-100 (PC) significantly upregulated TNF-α expression compared to the NC. Notably, TNF-α expression in the P4 treatment group was lower than that in the NC group and significantly reduced compared to the PC group. In terms of NF-κB expression, both p50 and p65 subunits showed decreased levels in the PC group relative to the NC group. The P1, P2, P3, and P5 treatment groups exhibited expression levels generally comparable to or slightly higher than those in the NC group. In contrast, the P4 group showed NF-κB p50 and p65 subunit expression levels that were comparable to or slightly lower than the NC group.
The E. coli challenge and phytobiotic supplementation resulted in significant changes in the expression of tight junction and mucin genes in the ileal and colonic mucosa of weaned piglets (Fig. 2).
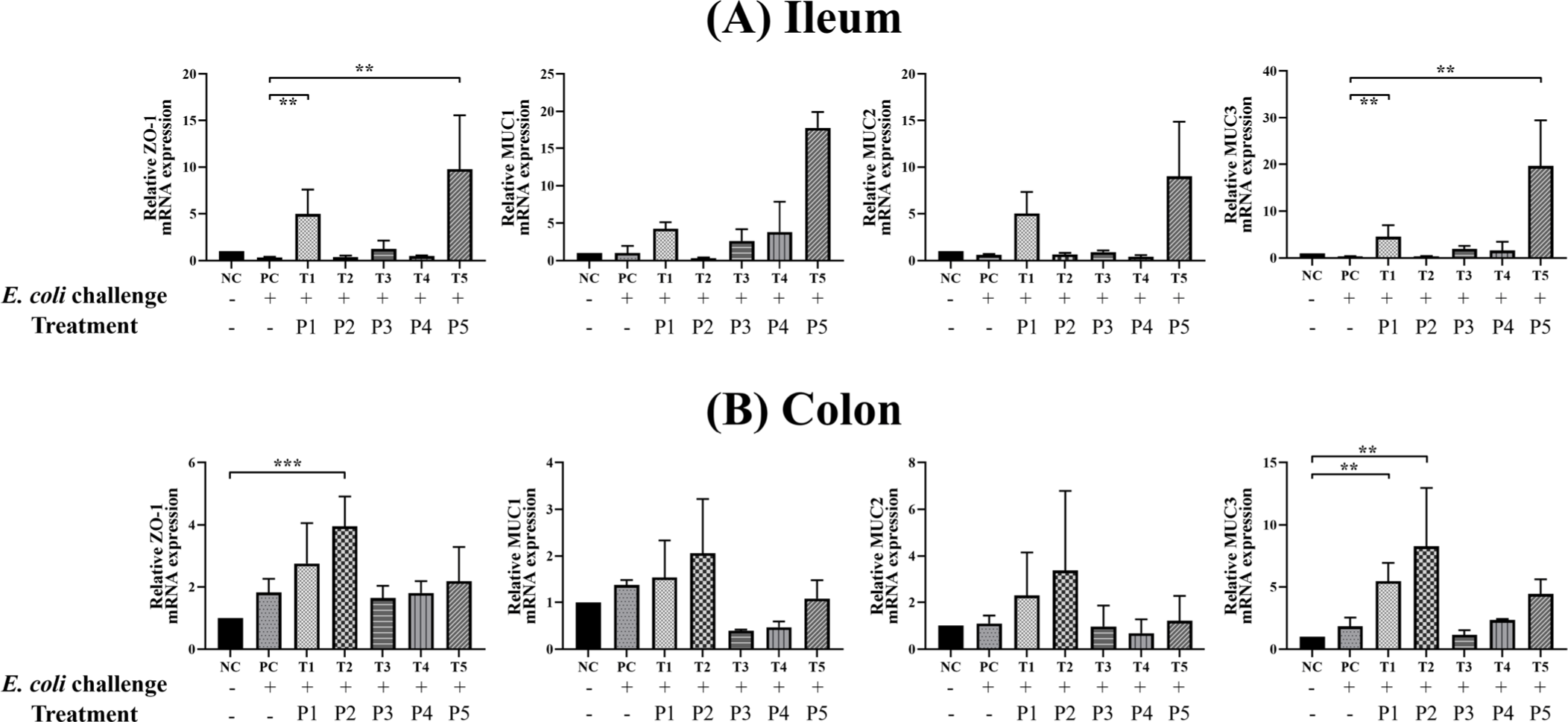
In the ileum, the PC group showed relatively lower expression of ZO-1, MUC2, and MUC3 genes compared to the NC group. In contrast, piglets in the T1 and T5 treatment groups exhibited significantly higher expression of ZO-1 and MUC3 genes compared to the PC group. In the colon, gene expression levels in the PC group were comparable to those in the NC group. However, the T2 group showed significantly higher expression of ZO-1, while both T1 and T2 groups demonstrated significantly elevated MUC3 expression compared to the NC group.
From the 42 samples, a total of 5,101,284 raw 16S rRNA gene sequence reads were obtained. After quality filtering, approximately 52% of the reads (2,662,821 reads in total) were retained for downstream analysis with per-sample read counts ranging from 13,738 to 154,970. These high-quality reads were used for microbial community analysis of the weaning piglets across experimental treatments.
Alpha diversity was assessed using Observed Features, Chao1, Shannon, and Simpson indices. No significant differences were observed in alpha diversity before (day 8) and after (day 21) the experiment regardless of the E. coli challenge or phytobiotic supplementation (Table 4).
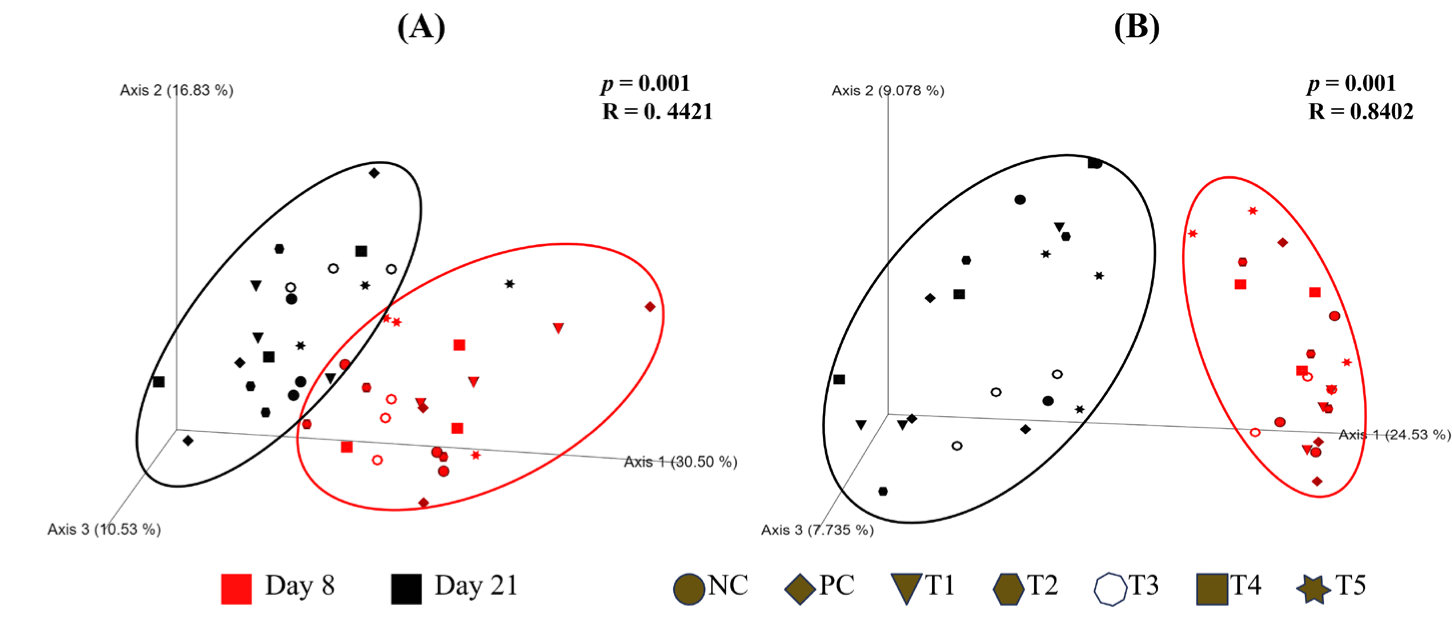
Beta diversity was analyzed using the ANOSIM to compare the weighted and unweighted UniFrac distances. PCoA was used to visualize group clustering (Fig. 3). The ANOSIM results based on unweighted UniFrac distances showed an R-value of 0.8402, indicating a distinct shift in the microbial community structure between pre-experiment (day 8) and post-experiment (day 21) samples. However, the PCoA plots based on both weighted and unweighted UniFrac distances showed that the microbial communities from pre-experiment (day 8) and post-experiment (day 21) samples were not clearly separated, indicating substantial overlap in community composition despite the observed structural differences.
We investigated the fecal microbial community composition of weaning piglets before and after the E. coli challenge and phytobiotic supplementation. Taxonomic assignment of ASVs was performed using the RDP database. At the phylum level, 14 phyla were identified (Fig. 4A). Bacillota was the most dominant phylum in all groups, constituting 43.85%–72.06% on day 8 and 60.24%–74.12% on day 21. Pseudomonadota significantly decreased from day 8 to day 21 in the NC (11.88% to 3.11%), PC (32.27% to 4.95%), T1 (18.41% to 0.76%), T2 (7.81% to 0.65%), and T4 (10.34% to 0.19%) groups. However, it increased in the T3 (5.55% to 7.49%) and T5 (6.47% to 15.67%) groups. Additionally, Actinomycetota significantly increased in the T3 group from 1.43% to 11.86%.
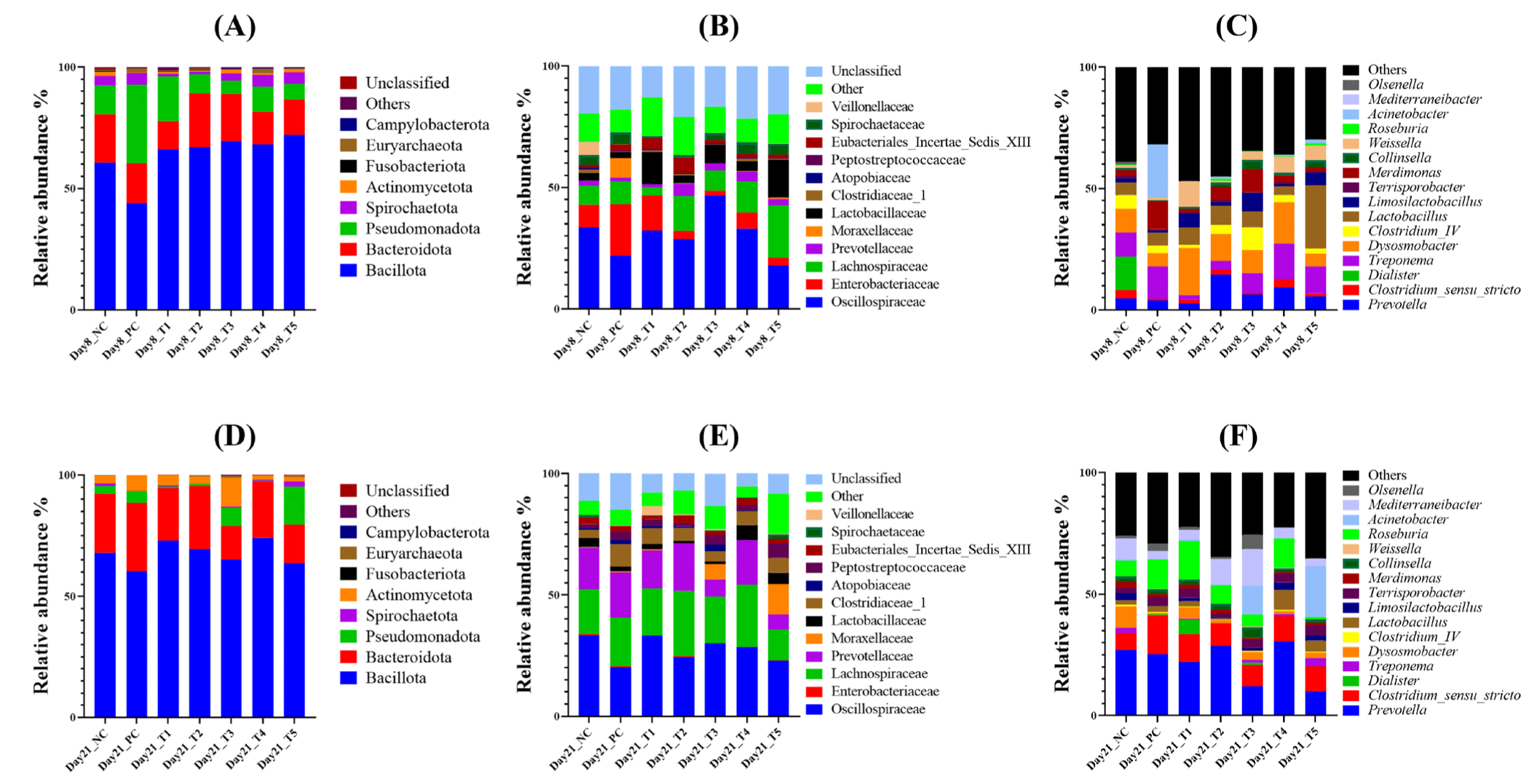
At the family level, 71 families were identified, with 12 predominant families identified and the others categorized as “Others” (Fig. 4B). On day 21, the most abundant families across all groups were Oscillospiraceae, Lachnospiraceae, Prevotellaceae, and Clostridiaceae_1. Prevotellaceae significantly increased in all groups, rising from 1.2%–5.14% on day 8 to 6.22%–19.52% on day 21. In contrast, Enterobacteriaceae, which ranged from 2.03% to 21% on day 8, decreased to only 0.01%–0.65% by day 21. Moraxellaceae significantly increased in the T3 (from 0.002% to 6.27%) and T5 (from 0.64% to 12.55%) groups.
At the genus level, microbial profiling revealed increased relative abundances of Prevotella and Clostridium sensu stricto in all groups by day 21, while Hydrogeniiclostridium and Treponema decreased (Fig. 4C). Lactobacillus showed a decrease in relative abundance from day 8 to day 21 in most groups, except for the T4 group, where it increased from 3.65% to 8.24%. Similarly, Limosilactobacillus increased in the NC group (from 1.57% to 3%). However, it decreased in the E. coli challenge groups (PC, T1, T2, T3, and T5), while increasing from 0.85% to 3.04% in the T4 group. Acinetobacter was predominant only in the T3 (11.79%) and T5 (21.01%) groups on day 21. Within the phylum Actinomycetota, the genera Olsenella and Collinsella were also identified.
Comparative analysis of genera on day 21 showed that the relative abundance of Lactobacillus was higher in the T4 and T5 groups compared to the NC and PC groups (Fig. 5A), with a significant increase in T4 (p < 0.05). Mediterraneibacter abundance was significantly lower in the PC group compared to the NC group, but it was significantly enriched in the T3 group (p < 0.05; Fig. 5B).
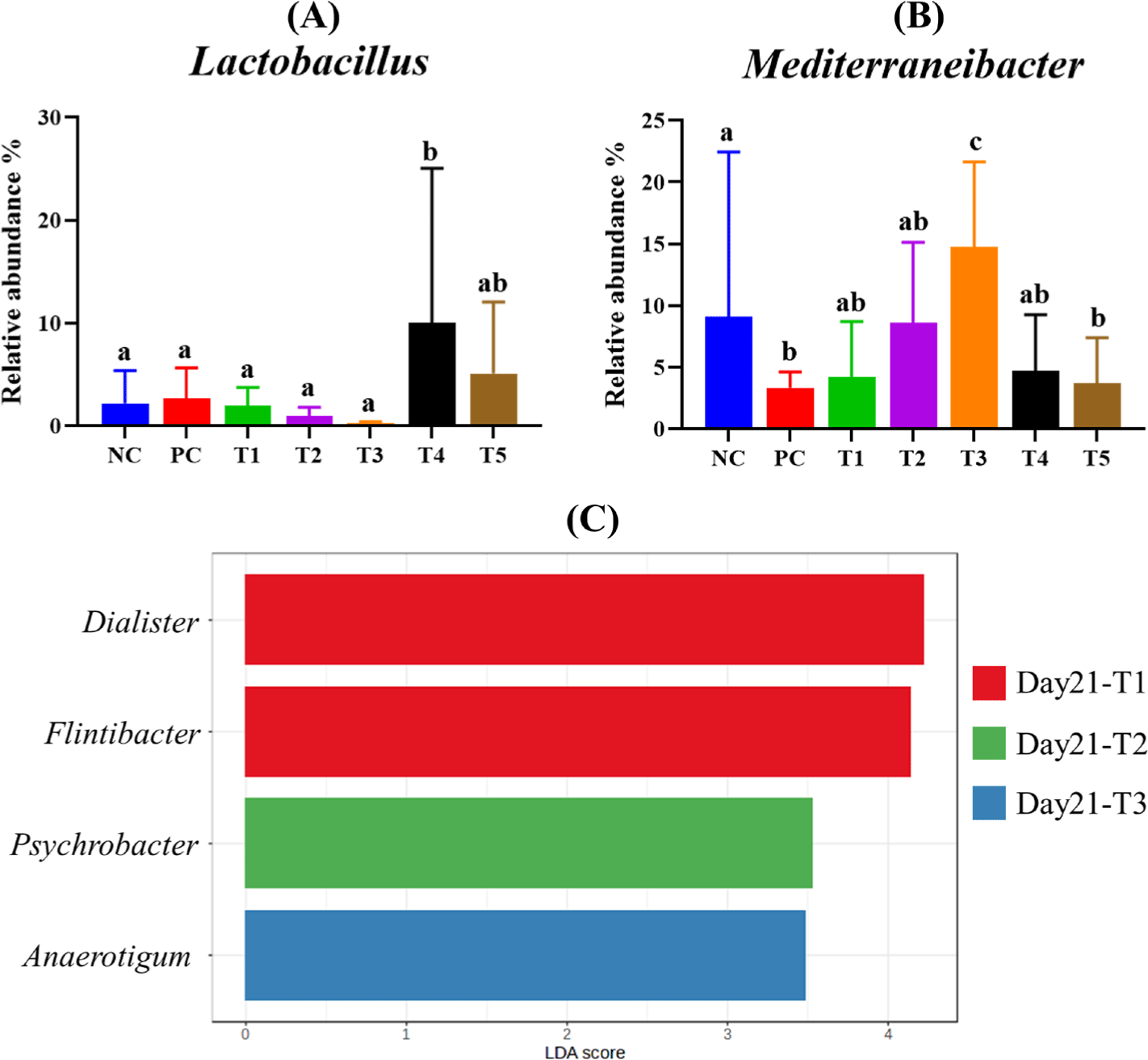
Linear discriminant analysis effect size (LEfSe) was conducted with a p-value cutoff of 0.1 and a Log LDA score of 3, and it identified four genera that were significantly enriched in each group on day 21 (Fig. 5C). Dialister and Flintibacter were characteristic of the T1 group, while Psychrobacter was representative of the T2 group. However, Anaerotigum was characteristic of the T3 group.
DISCUSSION
In this study, we investigated the cytotoxic and immunomodulatory effects of phytobiotics by analyzing the expression of TNF-α and NF-κB in RAW 264.7 cells treated with phytobiotics. Overall, the results showed that the tested phytobiotics did not exhibit cytotoxicity and might possess potential for modulating immune responses in weaning piglets.
The mucus layer, primarily composed of mucins such as MUC2 and MUC3, functions as a physical barrier that prevents direct bacterial contact with the epithelial surface, thereby limiting pathogen access and colonization [18,19]. Infection with enterotoxigenic Escherichia coli (ETEC) has been shown to compromise epithelial barrier function, resulting in electrolytes and water imbalances and the downregulation of protective mucosal proteins [20]. In our study, the expression of tight junction and mucin genes in the ileum and colon following ETEC challenge revealed that the ileum was more significantly affected. The observed reduction in the expression of tight junction proteins and mucin in the ileum of the PC is likely due to the preferential adhesion of ETEC fimbriae F18 to specific receptors present in the small intestinal epithelium [21,22]. This finding is consistent with previous reports by Gao et al. [23] and Becker et al. [24], which documented decreased expression of occludin and ZO-1 in the jejunum and ileum of pigs infected with ETEC. In this study, phytobiotic supplementation led to the upregulation of mucin and tight junction gene expression, suggesting a potential role in improving epithelial barrier function and maintaining intestinal homeostasis in weaned piglets. These results align with previous findings demonstrating the protective effects of phytobiotics against ETEC-induced intestinal damage. For instance, Liu et al. [25] reported that pigs infected with ETEC and supplemented with phytobiotics exhibited significantly greater villus height and elevated expression of tight junction-associated genes, which contributed to improved intestinal integrity. Similarly, Girard et al. demonstrated that dietary supplementation with chestnut extract rich in tannins reduced the incidence of diarrhea and enhanced growth performance in ETEC-infected pigs, further supporting the anti-inflammatory and barrier-protective properties of phytobiotics [12].
In this study, the diversity of the gut microbiota in weaning piglets generally decreased as weaning progressed, accompanied by a notable shift in microbial community composition. At the family level, a general increase in Lachnospiraceae and Prevotellaceae was observed, while Enterobacteriaceae decreased. These findings are consistent with other studies, which attribute these shifts to the transition from a milk-based diet to solid feed [26–28]. Lachnospiraceae and Prevotellaceae have been widely recognized for their crucial contributions to gut health [29,30]. Both families are prominent producers of short-chain fatty acids (SCFAs) including acetate and butyrate, which play essential roles in maintaining intestinal homeostasis. SCFAs serve as primary energy sources for colonocytes, promote mucosal immunity, and contribute to the regulation of inflammatory responses [31,32]. Moreover, SCFAs strengthen the intestinal barrier by enhancing tight junction integrity and lowering luminal pH, thereby creating an unfavorable environment for pathogenic bacterial colonization [33,34]. The observed increase in Lachnospiraceae and Prevotellaceae during the weaning period may thus reflect not only a microbial adaptation to dietary changes but also a favorable shift toward enhanced mucosal defense and resistance to enteric pathogens.
No significant differences in microbial diversity were observed between the NC group and the other groups challenged with E. coli. Additionally, comparisons of microbial composition revealed minimal differences in community structure between the PC and NC groups. This observation is consistent with previous studies reporting that ETEC exerts only a limited impact on the overall fecal microbial community structure during the post-weaning period [35,36].
It is well established that specific plant secondary metabolites found in phytobiotics can modulate gut bacterial communities by selectively promoting or inhibiting the growth of certain microbial taxa [37]. In our study, distinct shifts in bacterial composition were observed across treatment groups, suggesting compound-specific effects. Notably, the relative abundance of Pseudomonadota decreased more markedly in the T1, T2, and T4 groups compared to the NC and PC groups. In contrast, both Actinomycetota and Pseudomonadota were more prevalent in the T3 and T5 groups, indicating that the phytobiotic blends used in these treatments may favor the proliferation of these phyla.
In the phylum Pseudomonadota of T3 and T5 groups, the majority of the microbial composition was represented by the genus Acinetobacter. The genus Acinetobacter is typically recognized as an opportunistic pathogen associated with health-related infections [38–40]. However, several studies have reported that Acinetobacter species can inhabit the mammalian gut, although the ecological roles of strains other than the commonly studied pathogenic types remain largely unexplored [41]. While the precise mechanisms underlying their presence in the gastrointestinal tract are not fully understood, Acinetobacter spp. have been reported to participate in the degradation and metabolism of phytobiotics [42]. This suggests that the increased abundance of Acinetobacter observed in our study may be linked to the metabolic activity induced by the specific phytobiotic formulations administered in the T3 and T5 groups.
In the T3 group, the observed increase in the phylum Actinomycetota was attributed to the elevated abundance of the genera Collinsella and Olsenella. This increase may be associated with carbohydrate fermentation, potentially influenced by the excipients included in the T3 diet. Subramaniam et al. reported that various inactive pharmaceutical excipients, particularly those based on polysaccharides, can serve as fermentable substrates for gut microbes, thereby promoting microbial diversity and abundance [43]. Collinsella has been reported to produce SCFAs from both animal- and plant-derived carbohydrates such as lactose, fructose, and starch [44]. Similarly, Olsenella species are capable of fermenting carbohydrates and producing SCFAs including acetate, as metabolic by-products [45]. Beyond their SCFA production capacity, increases in Collinsella and Olsenella have been associated with elevated levels of IL-10, an anti-inflammatory cytokine involved in immune regulation and mucosal homeostasis [46]. These findings suggest that these genera may contribute to maintaining microbial diversity and ecological balance in the gut, thereby limiting pathogen colonization.
Lactobacillus was more abundant in the T4 and T5 groups compared to the NC and PC groups, with significantly higher levels observed in T4. The genus Lactobacillus is well known as a beneficial probiotic bacterium [47]. Its increase abundance has been associated with enhanced SCFA production, which can help prevent the invasion of pathogenic bacteria in the gastrointestinal tract while supplying energy to epithelial cells and strengthening gut barrier function [48,49]. Collado et al. [50] demonstrated that Lactobacillus can inhibit the adhesion of pathogens, including E. coli, to porcine intestinal mucus suggesting its potential role in suppressing E. coli colonization.
In addition, dialister was more abundant in the T1 group, and Psychrobacter was characteristic of the T2 group. Both genera have been reported as commensal gut bacteria commonly found in healthy pigs [51–53]. Yang et al. demonstrated that Psychrobacter may function as a probiotic, potentially contributing to increased gut microbial diversity [54]. Furthermore, the genus Flintibacter was more abundant in the T1 group. Flintibacter has also been shown to produce butyrate, a key SCFA involved in maintaining gastrointestinal health [55,56]. These findings indicate that the phytobiotics used in this study have the potential to beneficially modulate the gut microbiome of weaning piglets by promoting the growth of commensal and probiotic bacteria.
In summary, our results suggest that dietary supplementation with phytobiotics may enhance immune responses, mitigate inflammatory reactions, and beneficially modulate the gut microbiota in weaned piglets. However, further research is necessary to elucidate the specific roles and functional contributions of the microbial taxa influenced by phytobiotic supplementation, as many of these remain incompletely characterized in the context of gut health. In particular, the variation in microbial responses according to the duration of supplementation and the specific phytogenic compounds used warrants deeper investigation. A more comprehensive understanding of these temporal and compositional dynamics could support the development of optimized dietary strategies aimed at promoting microbial stability and improving host resilience during the critical weaning period.