
Background
Heat shock (or stress) proteins (HSPs) are a family of highly conserved cellular proteins that play an important role in protection from stress stimuli and metabolic insults in almost all organisms [1–4]. They include three major families: Hsp90 (85–90 kDa), Hsp70 (68–73 kDa) and low molecular-weight Hsps (16–47 kDa) [3]. The Hsp70 family is encoded by two different genes: a constitutive, “housekeeping” heat shock cognate (hsc) 70 gene, which is predominantly associated with physiological processes, and stress-inducible hsp70. Hsc70 protein plays a key role as molecular chaperone in a wide range of cellular processes, such as protein assembly, folding, transport through membrane channels, translocation and denaturation [4–7]. Hsp70 protein is mainly responsible for the maintenance of cellular homeostasis during the stress response, thus protecting cells from the damage caused by environmental stress agents, such as heat shock, chemical exposure and UV or γ-irradiation [4, 8, 9]. Hsp70 is also a potential activator of the innate immune system mechanisms [10–12]. Hsp70 is considered to be by far the most evolutionary conserved protein, found in all organisms from archaebacteria and plants to humans [13]. Both proteins have a modular structure consisting of a highly conserved N-terminal ATPase domain, an adjacent well-conserved substrate-binding domain (SBD) that contains a hydrophobic pocket with a lid-like structure over it, and a conserved but more variable C-terminal domain, which plays an important role in Hsp70 functions required for cell growth. In the ATP-bound state, the substrate-binding pocket is open and rapidly exchanges substrate. ATP hydrolysis induces closing of the lid over the pocket, which stabilises substrate binding. Return to the ATP-bound state restores the open conformation, facilitating substrate release [14, 15].
In aquaculture, fish are often exposed to stressful situations, such as sudden temperature changes, high stocking density, trauma, hypoxia, as well as viral and bacterial infections, which often results in high fish mortality. Both genes have been identified and their expression characterised in many fish species, e.g., rainbow trout (Oncorhynchus mykiss), zebrafish (Danio rerio), Korean rockfish (Sebastes schlegeli), Nile tilapia (Oreochromis niloticus), mandarin fish (Siniperca chuatsi) [16–20] and both are known to have a crucial role in response to heat shock, hypoxia, crowding stress and bacterial pathogens in fish [3, 4, 7, 19]. However, according to our best knowledge, a three-dimensional (3-D) model of any fish Hsp70 family protein has not been published so far.
Blunt snout bream (Megalobrama amblycephala Yih, 1955), native to the middle portions of the Yangtze River basin, is becoming an increasingly important freshwater aquaculture species in China. Due to its successful artificial propagation and high economic value, the total output of the blunt snout bream aquaculture industry reached 652 215 tons in 2010 [21–23]. Previously, Ming, Xie [7] used bioinformatics tools to analyse some physicochemical characteristics of the two Ma-Hsp70 family proteins, such as molecular weight, isoelectric point, solubility (as hydrophilic property) and richness in B cells antigenic sites. Their results indicated that Ma-Hsp70 shares more than 85 % identity with its homologs in other vertebrates, has no signal peptide or transmembrane region, contains many protein kinase C phosphorylation sites, N-myristoylation sites, casein kinase II phosphorylation sites and N-glycosylation sites, while the predominant elements of the secondary structure are α-helix and random coil. However, as the previous study left many questions regarding the physicochemical and structural properties (particularly regarding the tertiary structure) open, this study, as a successive work, aims to fill this gap. Several different computational tools and available web servers were used to deeply analyse the physicochemical characteristics and, using homology modelling, reliably predict the tertiary structure of the blunt snout bream Hsp70 and Hsc70 proteins. Additionally, rapidly increasing number of known gene sequences in many organisms has prompted the need for new procedures and techniques for the high-throughput functional annotation of genes. While most of those traditionally used remain rather costly and work-intensive, with rapidly growing number of protein structures deposited in the Protein Data Bank (PDB), computational structural genomics is becoming an increasingly promising tool for fast and cheap insight into protein structures, functions and interactions [24–27]. As Ming, Xie [7] analysed the expression of Ma-hsp70 and Ma-hsc70 genes in order to gain insight into their functions, this study further aims to provide a supplementary evidence for conserved function of these two genes by in-deep structure analysis and functional annotation of their polypeptide products on the basis of the similarity of their tertiary structures to the available templates from other organisms. As structure-based functional annotation has seldom been used in study of fish proteins, the aim of this study is also to test the applicability of this approach for functional annotation of fish gene sequences.
Methods
Amino acid sequences of the blunt snout bream Hsp70 (Accession number: ACG63706.2) and Hsc70 (Accession number: GQ214528.1) [7] were obtained from the NCBI protein database (http://www.ncbi.nlm.nih.gov/) in FASTA format as the target template and used for further analyses. Physicochemical properties of the proteins, including molecular weight, amino acid composition, theoretical isoelectric point (pI), the total number of positive and negative residues, extinction coefficient (EC), instability index (II), aliphatic index (AI) and grand average of hydropathicity (GRAVY) were analysed using Expasy’s ProtParam prediction server [28]. SOSUI server [29] was used to determine whether it is a soluble or a transmembrane protein, while CYS_REC (http://linux1.softberry.com) was used to predict the presence of cysteine residues and their bonding patterns.
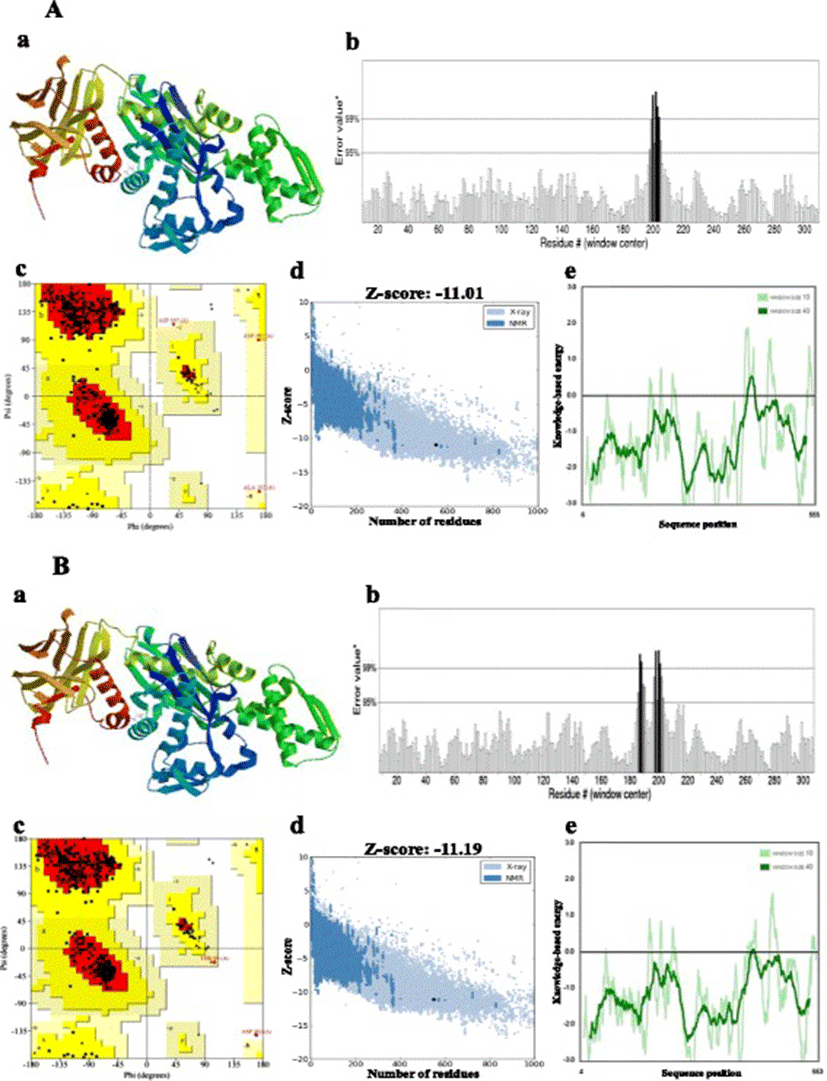
Homology modelling of the proteins was performed by the SWISS-MODEL server [30, 31], which aligns an input target with pre-existing templates to generate a series of predicted models. The most suitable template to build the 3-D model was selected on the basis of sequence identity [32]. Multiple amino acid sequence alignment was performed with ClustalW2 (http://www.ebi.ac.uk/Tools/msa/clustalw2). Stereochemical quality and accuracy of the predicted models were analysed using PROCHECK’s Ramachandran plot analysis, ERRAT, PROVE, Verify3D (all four available from the SAVES server at http://nihserver.mbi.ucla.edu), ProQ [33] and ProSA [34, 35]. Structural analysis was performed and model figures generated by Swiss PDB Viewer [36].
COFACTOR web server was used to perform the global structure match using TM-align algorithm and render the TM-score was calculated to assess the global structural similarity: values range from 0 to 1, where TM-score = 1 indicates the perfect match between two structures. Scores below 0.17 correspond to randomly chosen unrelated proteins, whereas a score higher than 0.5 implies generally the same fold [37]. Annotations on ligand-binding sites, gene ontology and enzyme commission were performed by the I-TASSER suite, which structurally matches the 3-D model of Ma-Hsp70 and Ma-Hsc70 to the known templates in protein function databases [38–40].
Results and discussion
In this study, several different computational tools and available web servers were used to deeply analyse the physicochemical characteristics and to reliably predict the tertiary structure of the blunt snout bream Hsp70 and Hsc70 proteins (using homology modelling). This research corroborated the previous predictions regarding the Ma-Hsp70 and Ma-Hsc70 protein length (643 and 649 amino acids), molecular weight (70517.7 and 71240.3 Da), theoretical isoelectric point (pI = 5.36 and 5.31) and the total number of negatively and positively charged residues (94 and 81 and 96 and 82, respectively) [7]. Among the new findings, the computed pI value indicated that the proteins are acidic (pI < 7) in character, implying that they can be purified on a polyacrylamide gel by isoelectric focusing. The calculated extinction coefficient (EC), which is in direct correlation with the cysteine, tryptophan and tyrosine content, of Ma-Hsp70 and Ma-Hsc70 proteins at 280 nm was 33725/33350 (assuming all pairs of cysteine residues form cysteines) and 35090/34840 (assuming all cysteine residues are reduced) M−1cm−1, respectively. The instability index (II) value was 33.68 (Ma-Hsp70) and 35.56 (Ma-Hsc70), implying that both proteins are stable (II < 40) [41]. Similarly, the aliphatic index (AI) of both proteins had very high value of 83.44 (Ma-Hsp70) and 80.23 (Ma-Hsc70), indicating stability over a wide temperature range [42]. The N-terminal of both proteins was methionine. Estimated half-life values were also the same for both proteins: 30 h in mammalian reticulocytes (in vitro), >20 h in yeast (in vivo) and >10 h in Escherichia coli (in vivo). The grand average hydropathicity (GRAVY) of Ma-Hsp70 and Ma-Hsc70 was −0.431 and −0.473, respectively, corroborating that proteins are hydrophilic and highly soluble in water. Amino acid composition analysis revealed high amounts of alanine (8.4 %), lysine (8.2 %) and glycine (8.1 %) in Ma-Hsp70, whereas glycine (8.5 %), lysine (8.2 %) and aspartic acid (7.6 %) were the most abundant in Ma-Hsc70 (Table 1).
N represents the total number and % the numeric percentage of each amino acid
Though six and four cysteine residues were found in Ma-Hsp70 and Ma-Hsc70 sequences, respectively, no evidence was found for the existence of disulphide bonds, which are essential for the folding of proteins and responsible for stabilisation of protein structure [43] (Table 2).
Bovine Hsc70 (PDB ID: 4 fl9.1.A) at 1.9 Å resolution was chosen as the best available template to build a 3-D model for both Ma-Hsp70 (90.24 % sequence identity) and Ma-Hsc70 (95.85 % seq. identity) proteins using homology modelling. Template-target sequence alignments and 3-D structure of the predicted models are shown in Figs. 1 and 2, respectively. Verification of the results, using different tools, invariably indicated a good quality of the proposed models (Table 3). So the Ramachandran plot analysis, where a good model would be expected to have over 90 % of residues in the most favoured regions, suggested a good quality of both homology models (Ma-Hsp70 - 92.7 % and Ma-Hsc70 - 92.4 %). The overall G-factor of the two models, where a value > −0.5 indicates a good model [44], was 0.16. Verify3D analysis of the models revealed that 92.55 % (Ma-Hsp70) and 92.36 % (Ma-Hsc70) of the residues had an average 3D-1D score ≥0.2, while 98.36 % and 99.82 % were >0, respectively. As the cut-off score was ≥0, this implies the predicted models are valid [45]. Overall ERRAT quality factor value, expressed as the percentage of the protein for which the calculated value falls below the 95 % rejection limit, was 95.547 % and 92.963 %, respectively (Fig. 2). Good high resolution structures generally produce values around 95 % or higher [46]. LGscore and MaxSub index indicated “very good” (value >5.0) and “correct” quality (value >0.1) [33] for Ma-Hsp70 and Ma-Hsc70 protein models, respectively (Table 3). Z-scores for Ma-Hsp70 (−11.01) and Ma-Hsc70 (−11.19) models were within the range of scores typically found for the native proteins of similar size, while the plot of residue energies, where positive values correspond to problematic or erroneous parts of the input structure, revealed that most of the calculated values were negative [35] (Fig. 2). All of these validation tools strongly suggested that both proposed 3-D models could be accepted as reliable with high confidence.
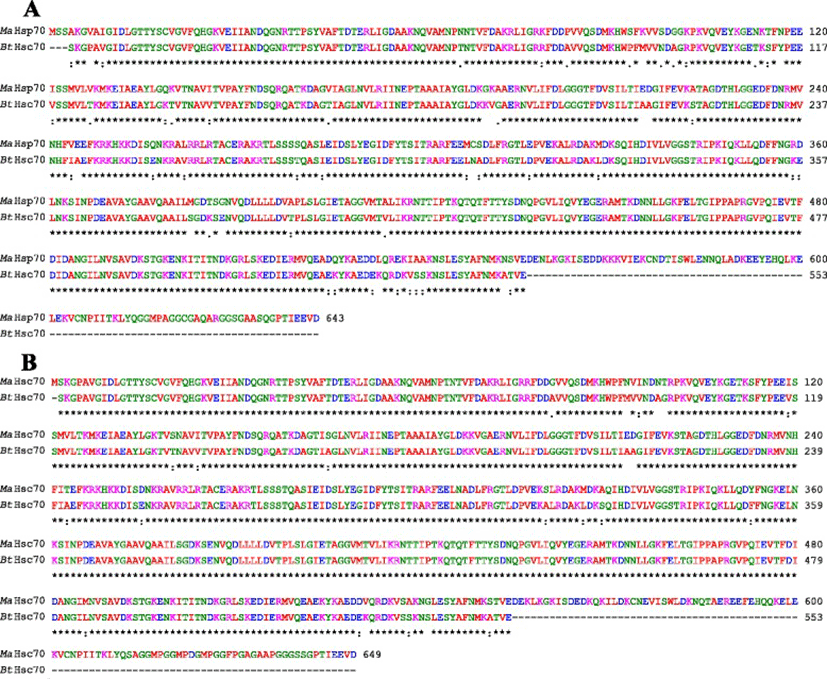
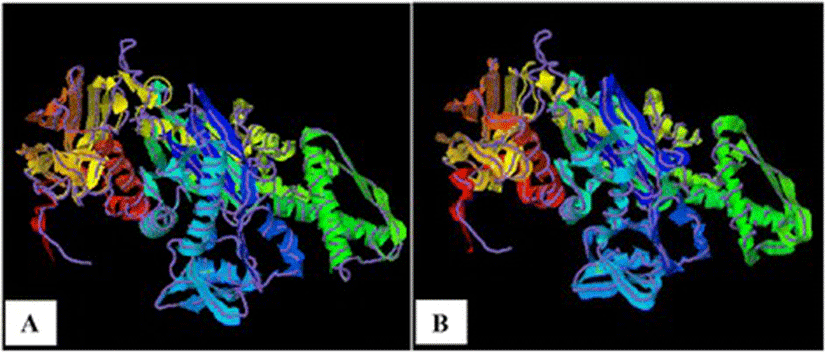
As TM-scores >0.5 indicate that two proteins generally have the same fold, the results (TM > 0.66) implied a very high level of structural conservation between Ma-Hsp70 and related rat, mouse, human and yeast proteins (Table 4). TM-score value of 0.986 between both Ma-Hsp70 and Ma-Hsc70 and Bos taurus Hsc70 3-D protein models indicates that they are structurally almost identical (Fig. 3). Somewhat surprisingly, TM-scores were identical for both protein models, while RMSDa IDENa and Cov. values were different between the two models. This could be explained by the fact that their tertiary structure is more similar than their primary and secondary structure, as well as by the absence of fish Hsp/Hsc70 templates in the PDB. The fact that the same PDB protein models (three Hsc and two Hsp) were indicated as the best available templates for both tested models reflects a very high level of conservation among the two proteins and other vertebrate Hsp70 family proteins (Ma-Hsp70 - 86 % identity and Ma-Hsc70 - 93 %), as well as between Ma-Hsp70 and Ma-Hsc70 (86.5 % identity) [7]. High structural similarity to Hsp70 protein family members from a wide range of taxonomically distant organisms additionally corroborated a remarkably high level of evolutionary conservation among the members of this protein family and provided an indirect evidence for a high level of functional conservation as well.
Analogs were inferred by COFACTOR analysis, based on the TM-score of the structural alignment between the query structure and known structures in the PDB. RMSDa is the average root mean square deviation between residues that are structurally aligned by TM-align; IDENa is the percentage sequence identity in the structurally aligned region; Cov. represents the coverage of the alignment by TM-align and is equal to the number of structurally aligned residues divided by length of the query protein
An ATP-binding site was predicted with a very high confidence, on the basis of structure similarity with bovine Hsc70 (PDB ID = 1kax-A; C-score = 0.98) and yeast actin (1yag-A; 0.96) for Ma-Hsp70 and Ma-Hsc70, respectively. High structural identity with yeast actin [47] reflects structural and mechanistic similarities between ATP hydrolytic mechanisms in proteins with different functions. In line with its ATPase function and previous findings [48], a phosphate ion (PO4)-binding site was also predicted with a relatively significant confidence on the basis of structural similarity with the bovine Hsc70 II (PDB ID = 1hpm-A; C-score = 0.22) for Ma-Hsp70 and the N-terminal domain of the Cryptosporidium parvum Hsp70 (PDB ID = 3l4i-B; C-score = 0.23) for Ma-Hsc70 (Table 5).
Predicted by COACH analysis, based on the C-score of the structural alignment between the query structure and known structures in the PDB. C-score is the confidence score of the prediction, where a higher score (min-0, max-1) indicates a more reliable prediction; Clust. size is the total number of templates in a cluster; ligand is the name of a possible binding ligand
In accordance with the predicted ligands, Enzyme Commission analysis (performed by I-TASSER program) predicted, with relatively similar confidence scores, both Ma-Hsp70 and Ma-Hsc70 to be either of the isozymes hexokinase and glucokinase, both of which can transfer an inorganic phosphate group from ATP to a substrate. Somewhat confusingly, deoxyribonuclease function was also predicted for both proteins (Table 6), however, upon closer inspection of the 3cjc PDB entry, it became obvious that this prediction was the result of an error in the data retrieval from the PDB, as the 3cjc-A chain represents actin, which is an ATPase, with an adenosine-binding site, while deoxyribonuclease is the 3cjc-D chain in the entry.
CscoreEC is the confidence score for the enzyme commission number (EC No.) prediction (0–1), TM-sc is the TM-score, Cov represents the coverage of global structural alignment and is equal to the number of structurally aligned residues divided by length of the query protein. See Table 4 for other term explanations
Similarly, consensus prediction of GO terms also suggested ATP binding, interacting selectively and non-covalently with adenosine 5'-triphosphate (GO = 0005524; ontology = molecular function) as the main function for both proteins, with very high GO-scores (0.98 for Hsc70 and 0.99 for Hsp70).
All these results are in accordance with the previously described functioning mechanisms: Hsp70s in the ATP-bound state catch and release their substrates rapidly, while Hsp70s in the ADP-bound state seize them firmly. By cycling between the ATP- and ADP-bound states, Hsp70s exert their chaperone activity [14, 49]. However, the analysis was not sensitive enough to distinguish between the functions of the constitutive (Hsc) and inducible (Hsp) isoforms. This is not a major setback, as it has been suggested that the functional difference appears to lie more in regulation of the SBD-substrate interactions than in the physical properties of the two ATPase domains [15].
Conclusions
To help better understand the functional biology of Hsp70 and Hsc70 in blunt snout bream, several computational tools were used to analyse the physicochemical properties, generate valid homology models of both proteins and predict their functions on the basis of structural similarity to other protein templates. Apart from presenting the first published homology models of Hsp70 and Hsc70 proteins in fish, this research also revealed a high structural similarity to Hsp70/Hsc70 proteins from several taxonomically distant animal species, corroborating a remarkably high level of evolutionary conservation among the members of this protein family. Functional annotation based on structure similarity provides a reliable additional indirect evidence for a high level of functional conservation of these two genes/proteins in blunt snout bream, but it is not sensitive enough to distinguish between the two isoforms. In conclusion, even though gene function assignment based on protein structure similarity is at present somewhat limited by the number of available protein structures deposited in the PDB, it has a strong potential to become a very fast, cheap and relatively reliable technique for high-throughput gene function assignment in fish.