
Background
The Sapsaree (Canis familiaris) is a native Korean dog that is distributed throughout the Korean peninsula, and is very friendly, protective, and loyal to its owner. The Sapsaree population size decreased dramatically during the Korean War in the 1950s, and the breed was considered endangered. A program of systematic mating and reproduction to preserve the Sapsaree from extinction and maintain a pure pedigree generated the current population of about 4,000 individuals, including the 500 dogs now living at the Sapsaree Breeding Research Institute in Gyeongsan, Gyeongbuk province [1]. The Sapsaree was registered as a Korean Natural Monument (number: 368) in 1992. Although recent studies have shed light on the origin and various morphological and behavioral traits of the Sapsaree, the genetic backgrounds, abundant genetic polymorphisms, and novel genes of the Sapsaree are still not completely understood [2].
RNA sequencing (RNA-Seq) is one of the most useful next-generation sequencing tools for investigating the landscape of a whole transcriptome. Using RNA-Seq, researchers have identified differentially expressed and novel genes, unraveled the expression profiles underlying phenotypic changes, and discovered unannotated, transcriptionally active regions that cannot be detected by conventional gene prediction [3]. Furthermore, the recent use of RNA-Seq has captured the scale and complexity of organ-specific and tissue-specific transcriptomes, making RNA-Seq the technique of choice for investigating gene expression during complex phenomena such as stress [4].
Exercise is usually recognized as a stress factor, as is any environmental change that activates or pressures cells and tissues. And the level of response to physical exercise is different from individuals to individuals. So, it is needed to research the biological responses under physical stress. Several RNA-Seq studies of exercise-induced stress have been performed in equines [3, 4]. There has been little progress, however, in canine-based RNA-Seq studies, which have been limited to certain diseases. Therefore, we performed a large-scale analysis of whole-transcriptome data to investigate the gene expression levels before and after exercise in the Sapsaree. This study will provide the basic approach for identified the biologic characteristics and breeding the working dogs.
Methods
The dogs were handled in accordance with Article 23 ‘Experiments with Animal’ of Korea’s Animal Protection Law, 2015, and the Korean Sapsaree Foundation cooperated and approved all animal care procedures prior to the initiation of the experiment. Ten Sapsarees housed in controlled environmental conditions were used in the experiment. The basic morphological traits of each Sapsaree including body height (BH), body length (BL), depth of chest (DC), body weight (W), and hair color were measured before the experiment. Then, the dogs were exercised one-on-one by trainers for 1 h in 5 min intervals, with the trainers leading the dogs in quick step 10 times around a course on a 20 m × 20 m square field located at the Korean Sapsaree Foundation facility in Gyeongsan. The exercise course included hurdling (40 cm, 70 cm, and 80 cm), high jumping (50 cm), A-shape hurdling, bridge-shape hurdling, and seesaw hurdling.
Blood samples were drawn under a veterinarian’s supervision from a cephalic vein before and immediately after exercise, resulting in a total of 20 samples. Immediately after collection, portions of the blood samples were dispensed into serum separator tubes (SST) (1.5 ml in each tube [Greiner Bio One, Kremsmuenster, Austria]) to measure hormone levels, and into PAXgene blood RNA tubes (2.5 ml in each tube, [PreAnalytiX, Hombrechtikon, Switzerland]) for RNA sequencing according to the respective manufacturers’ protocols [5, 6]. The blood for serum was allowed to clot at room temperature before centrifugation. The serum was then separated and stored frozen at -70 °C until completion of the case enrollment and sample collection.
Four substances related to physical stress were chosen based on previous studies: cortisol, aspartate aminotransferase (AST), creatine kinase (CK), and creatinine. The serum CK, AST, creatinine levels are commonly used to muscle damage indicators and renal function [7–10]. And a recent study indicated that physical stress resulted in immediate increase in the plasma concentrations of cortisol [11]. The level of target substances in serum from the upper side of the SST tubes were measured using a BS-400 chemistry analyzer (Mindray, Shenzhen, China) and an Immulite 1000 Immunoassay System (Siemens, New York, USA) [12, 13].
Total RNA was extracted from the PAXgene tubes according to the manufacturer’s instructions [6]. Starting with the total RNA, mRNA was purified using poly (A) selection or rRNA depletion, converted into double-stranded cDNA, and amplified by PCR. To check the RNA quality, all the RNA samples were examined for RNA Integrity Number, and 28S to 18S rRNA value using Bioanalyzer. Next, the construction of library were used with the Illumina TruSeq RNA Sample Preparation Kit v2 (catalog #RS-122-2001, Illumina, San Diego, CA) following the manufacturer’s instructions [14]. And the library was quantified using the KAPA library quantification kit (Kapa Biosystems KK4854) following the manufacturer’s instructions [15]. The final individual libraries were sequenced using the Illumina Hiseq2000 platform, which created 100 bp paired-end (PE) RNA-sequencing reads.
To collect high-quality transcriptome data, we filtered the sequencing data by phred score (Q ≥ 20) and minimum length (≥25 bp) using the SolexaQA software [16]. The filtered reads were mapped to 48,370 reference mRNAs from Canis lupus familiaris using the bowtie2 software (mismatches ≤ 2) [17, 18]. The number of mapped reads for each mRNA was counted and then normalized using the DESeq packages in R [19]. Differentially expressed genes (DEGs) were selected by over 100 mapped read counts, a ≥ twofold change in reads coverage and a binomial test with a false discovery rate (FDR) ≤ 0.01 at the first. And then, the final DEGs were identified by a ≥ twofold change in reads coverage, a binomial test with a false discovery rate (FDR) ≤ 0.01, and a read count ≥ 1,000 either before or after exercise. The FDR was applied to identify the threshold p-value for multiple tests and was calculated using DESeq. Correlation analysis and hierarchical clustering was performed to group the genes according to patterns of expression using the AMAP library in R [20].
And we tested the stability of the gene expression levels before and after the exercise of housekeeping genes, such as HNRNPH1, GAPDH, RPL8, TAF4B, and TAF1 with t-test method supporting the statistical significance [21].
Functional enrichment analyses were carried out using the Gene Ontology (GO) database and including all three GO categories (biological processes, cellular components, and molecular functions), providing a structured and controlled vocabulary to describe the gene products [22]. We also used the KEGG database to identify the biological mechanisms and metabolic pathways associated with the differentially expressed genes corresponding their enzyme commission numbers [23]. DAVID is a web-accessible annotation system (https://david.ncifcrf.gov/home.jsp) that provides a comprehensive set of functional annotation tools for investigators for understanding the biological meanings behind large lists of genes [24]. We used DAVID to analyze the clusters of differentially expressed genes annotated by the Entrez gene IDs of the genes with counts ≥ 2 and FDR ≤ 0.1 of each GO and KEGG term.
To identify correlations between the gene expression pattern and body weight, we carried out t-tests comparing W/BH, W/BL, and W/DC values between two groups of dogs that showed different gene expression patterns. To control for group differences in baseline weight, the weight was divided by the BH, BL, and DC. A p-value ≤ 0.05 was used as the cutoff for significance in all analyses using the t-test function of R.
Results and discussion
The basic morphological traits of the 10 Sapsarees measured before the experiment are shown in Table 1. All the dogs were males, 13 to 60 months of age. The BH measured from the ground to the top of the withers ranged from 49 cm to 62 cm. The BL measured from the point of the shoulder to the rear point of the croup ranged from 58 cm to 70 cm. The DC measured from the elbow to the top of the withers ranged from 21 cm to 28 cm. The W measured by weighing balance ranged from 18.7 kg to 30 kg [25].
aMonths of age measured from birth to the date of exercise
bHair color : BT (Black & Tan), W (White), Y (Yellow), DY (Dark Yellow), SY (Strong Yellow)
cBH Body Height, dBL Body Length, eDC Depth of Chest, fW Body Weight
Compared with those before exercise, the concentrations of AST, CK, and creatinine were slightly increased after exercise, but the increases were not significant (Additional file 1: Tables S1 and S2). Cortisol, a key hormone from the adrenal glands, was significantly elevated after exercise in all individuals except for Cheongbaek (Additional file 2: Figure S1, Additional file 1: Table S2). Hence, it seems that cortisol can be used as a marker of exercise-induced stress.
After the RNA quality check, the RNA sequencing generated over 75 Gbp (about 3.8 Gbp per sample) of data consisting of 100 bp paired-end reads (Table 2). Trimming resulted in reads with a mean length of 86.58 bp across all samples and a total combined length of about 57 Gbp, which was 74.9 % of the raw sequence.
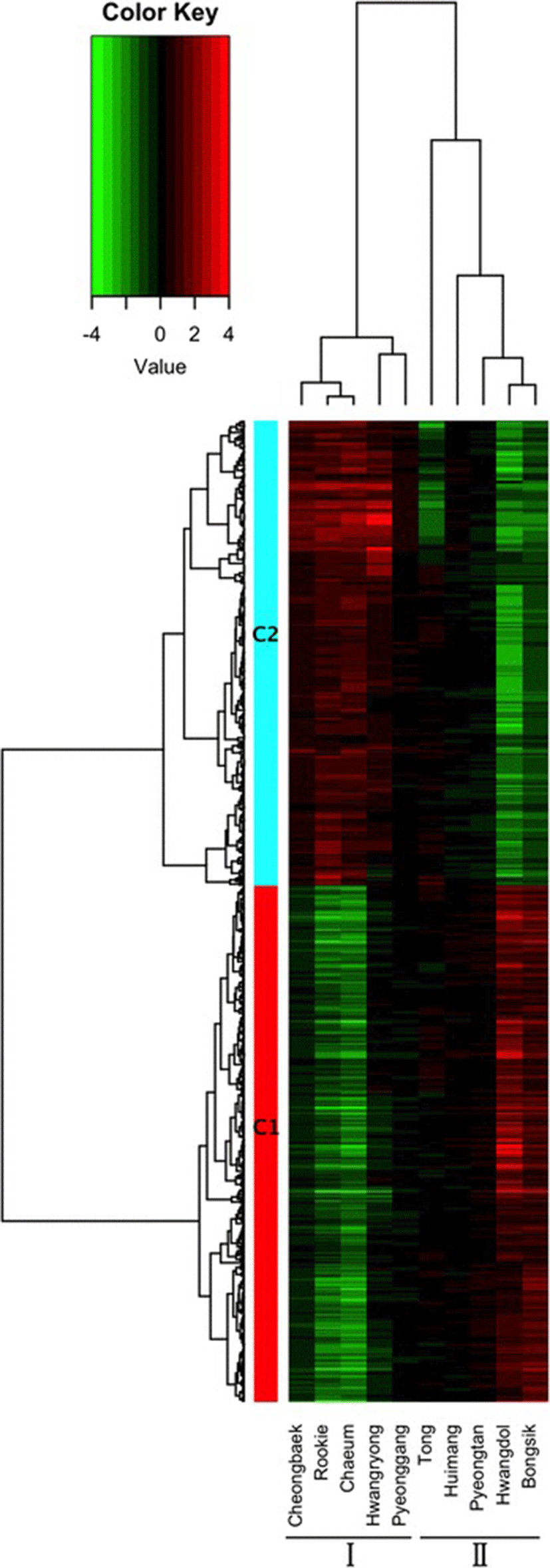
Using bowtie2, 80.76 % of the filtered reads were successfully mapped to the current dog reference genes (Canis lupus familiaris, 48,370 mRNAs) [18, 26]. A novel bioinformatics pipeline for processing large amounts of transcriptome sequences was built. We calculated the expression levels of all the genes with mapped reads from the 10 individuals before and after exercise. By comparing the coverage before and after exercise, we identified 2,549 DEGs. The numbers of up-regulated and down-regulated genes were different in each individual. Pyeonggang, Huimang, and Pyeongtan had fewer than 10 DEGs, but each of the other dogs had more than 100 genes (Additional file 3: Figure S2). After filtering out genes with low levels of expression, 525 genes were grouped into two clusters (C1 and C2) of 276 and 249 genes, respectively, depending on their expression pattern (Fig. 1 and Additional file 4).
And the reliability of the gene expression analysis based on RNA-Seq was confirmed by t-test which supported the statistical significance of the stability of housekeeping genes expression before and after the exercise (Additional file 1: Table S3).
A total of 26 unique genes among the 525 differentially expressed genes were assigned to 11 functional groups based on GO assignments (Additional file 1: Table S4). The genes were involved in biological processes such as programmed cell death, negative regulation of cell motion, regulation of cellular component biogenesis, protein metabolic process, and others. The cellular components linked to the genes were the intracellular parts, such as cytoplasm. The molecular function assignments were mainly to the catalytic and binding activities, such as cation binding.
A further functional classification of all the differentially expressed genes was performed using the KEGG database. A total of 84 unique genes among the 525 differentially expressed genes were assigned to 30 metabolic pathway terms, including phosphatidylinositol signaling system, ribosome, proteasome, oxidative phosphorylation, and others (Additional file 1: Table S5). The functional annotation analyses indicated that the genes regulated under exercise-induced stress were mainly related to the metabolite pathways.
Based on the gene expression patterns induced by exercise, the 10 Sapsaree individuals were divided into two groups (Groups I and II; Fig. 1). Group I included Cheongbaek, Rookie, Chaeum, Hwangryong, and Pyeonggang, while Group II included Tong, Huimang, Pyeongtan, Hwangdol, and Bongsik. The differentially expressed genes formed two clusters. Cluster 1 (C1) was down-regulated after exercise in Group I but up-regulated after exercise in Group II. Cluster 2 (C2) was up-regulated after exercise in Group I but down-regulated after exercise in Group II.
We examined the cortisol change after exercise, age, and body type for differences between the two groups. Only the body type showed a significant difference between Group I and Group II. Using W/BH, W/BL, and W/DC to characterize the overall body type, we found that Group I had a heavier body type than Group II (Table 1). In addition, W/BH, W/BL, and W/DC were each significantly different (p ≤ 0.05) between the two groups based on independent t-tests (Table 3). Those findings suggest differences in gene regulation between heavier dogs and lighter dogs under exercise stress.
| Mean of group Ib | Mean of group IIc | td | dfe | p-valuef | |
|---|---|---|---|---|---|
| W/BHa | 0.357 | 0.452 | -3.386 | 8.124 | 0.0093 |
| W/BLa | 0.312 | 0.397 | -3.211 | 9.413 | 0.0100 |
| W/DCa | 0.823 | 1.018 | -2.598 | 8.364 | 0.0306 |
aW Body Weight, BH Body Height, BL Body Length, DC Depth of Chest
bMean of Group I: average value of Group I
cMean of Group II: average value of Group II
dt : t-value, edf : the degree of freedom value, fp-value : significance
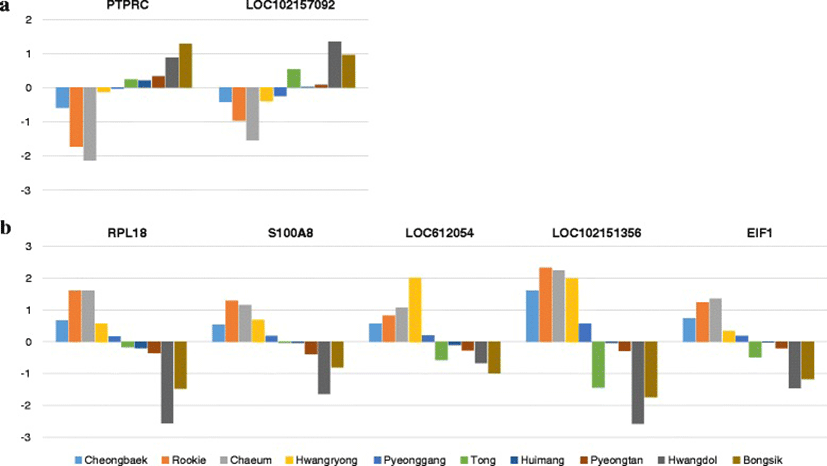
We identified 525 genes with differential expression before and after exercise. We manually curated seven genes (PTPRC, LOC102157092, RPL18, S100A8, LOC612054, LOC102151356, EIF1) that had distinctly different expression patterns before and after exercise in Groups I and II (Fig. 2). For example, PTPRC (Entrez ID : 490255) is described as ‘protein tyrosine phosphatase, receptor type, C’. Some genes had uncharacterized functions. Figure 2 shows the log2 fold changes of the seven selected genes after exercise for each of the dogs. There were clear differences in the changes in expression levels of the seven selected genes after exercise between the two groups of dogs. The seven selected genes could be used as markers of exercise stress and for grouping dogs according to body type.
Conclusions
This study provides the Sapsaree whole-transcriptome sequence data and identifies genes that are differentially expressed before and after exercise in the Sapsaree, which could be used as markers of exercise stress. The pattern of changes in global gene expression induced by exercise was different depending on the body type. RNA sequencing and gene expression analysis can be useful for grouping the Sapsarees by body type and response to exercise. This study provides a basis for future research investigating the biologic characteristics of the Sapsaree and the large-scale genomic differences of canines in general.