
INTRODUCTION
Herbivorous animals can utilize fibrous carbohydrates as feed resources through the actions of various carbohydrate hydrolyzing enzymes secreted by microorganisms in their gastrointestinal tracts (GIT) including rumen for foregut fermenters and cecum for hindgut fermenters. Produced volatile fatty acids and microbial cells are absorbed and used as energy and nitrogen sources by host animals. The performance and production of herbivorous animals are closely related to the microbial ecosystem and individual microorganisms in GIT and this has made scientists isolate, identify and characterize microorganisms from GIT. In the GIT of herbivores, bacteria, protozoa and fungi are the major microorganisms which degrade fibrous feed and bacteria and protozoa degrade fiber only with the reaction of fiber degrading enzymes [1,2]. However, the anaerobic fungi (AF) degrade fiber through their physical and biochemical reactions. During fiber degradation, the anaerobic fungi penetrate their rhizoid filaments into the fiber matrix [3] and secrete a wide variety of both carbohydrate hydrolyzing enzymes and protein hydrolyzing enzymes [4]. The carbohydrate hydrolyzing enzymes secreted from the anaerobic fungi during fiber degradation are both glucanases [5] and xylanases [6] and the former hydrolyzes cellulose or starch to hexose glucose and the later hydrolyzes hemicellulose to pentose xylose.
More than four decades, the carbohydrate degrading enzymes from the anaerobic fungi were considered as promising resources both for animal production and other industries including food, paper, fabric and bioenergy. For this reason, many research teams have tried to isolate and identify the anaerobic fungi either from the foregut or hindgut of herbivores. However, only limited numbers of anaerobic fungi have been isolated and identified from the gastrointestinal tract of herbivores due to the difficulties in culture conditions. Besides, the sequence analysis of anaerobic fungi had been hindered with their high adenine (A) and thymine (T) contents and high A–T rich repeats in nucleotide sequences. Recently, the development of high throughput sequencing using the next generation sequencer (NGS) made it possible to analyze the fungal genome and transcriptome with or without culturing technique. In this paper, the researches on the anaerobic fungi during the last four decades were reviewed based on publicly available DB.
ABOUT ANAEROBIC FUNGI
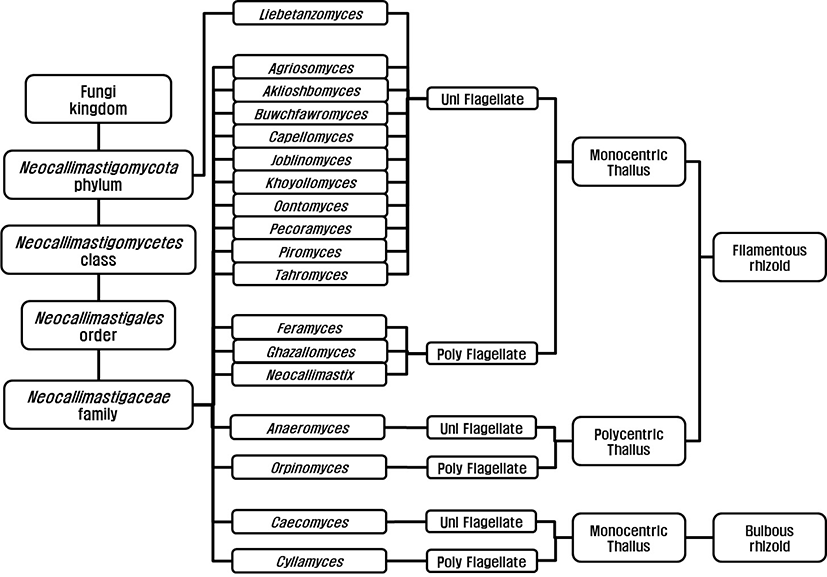
Anaerobic fungi were classified as flagellated protozoa due to their flagellated zoospore [7,8] before Orpin’s report (1975) and they were reclassified as family Neocallimastigaceae [9], order Neocallimastigales [10], and phylum Neocallimastigomycota [11]. Finally, the anaerobic fungi are classified in the phylum Neocallimastigomycota containing one class; Neocallimastigomycetes, one order; Neocallimastigales, one family; Neocallimasticaceae and 18 genera (Fig. 1). The genera in the family Neocallimasticaceae have been classified based on the morphological characteristics including zoospore flagellation (uniflagellate vs. polyflagellate), the sporangia development (monocentric vs. polycentric) and the thallus morphology (filamentous vs. bulbous) [12]. The number of flagella on zoospores is less than 4 in uniflagellate fungi and uniflagellate fungi are Agriosomyces, Akiloshbomyces, Anaeromyces, Buwchfawromyces, Caecomyces, Capellomyces, Joblinomyces, Khoyollomyces, Liebetanzomyces, Oontomyces, Pecoramyces and Piromyces. The polyflagellate fungi including Cyllamyces, Feramyces, Ghzallomyces, Neocallimastix, and Orpinomyces have more than 4 flagella on their zoospores. In monocentric fungi including Agriosomyces, Akiloshbomyces, Buwchfawromyces, Caecomyces, Capellomyces, Cyllamyces, Feramyces, Ghzallomyces, Joblinomyces, Khoyollomyces, Liebetanzomyces, Neocallimastix, Oontomyces, Pecoramyces and Piromyces, single sporangium containing a nucleus develops on its thallus and their rhizoidal system are anucleate. On the other hand, several sporangia develop on their thallus and the nucleus migrate through rhizoid with repeated division in polycentric fungi including Anaeromyces and Orpinomyces. The genera Caecomyces and Cyllamyces are bulbous type thallus with holdfast sporangia [13,14]. However, there have been many difficulties in morphology based identification due to variations in the number of flagellate and shape and size of sporangia under different nutritional environments [13]. The development of molecular technique led to the application of gene sequence for fungal identification and several different barcode markers were suggested. Dore and Stahl [15] compared the small subunit (SSU) rRNA genes of different anaerobic fungi for the first time, however, inter-relationship between genera was not clear due to highly conserved sequences in SSU. The internal transcribed spacer 1 region (ITS1) of rRNA was suggested by Li and Heath [16] and established [17]. Later several regions within the ITS1 were suggested for identification and quantification of anaerobic fungi [18,19]. The use of large subunit (LSU) rRNA gene was reported by Hausner and colleagues [20] for the first time and practical application methods using D1/D2 region within LSU for identification were reported [21,22]. Currently, both ITS1 and LSU sequences are analyzed for the identification of anaerobic fungi to compensate for the limitations of each marker [23].
The identified 35 species of anaerobic fungi were isolated from either ruminant (cattle, buffalo, sheep, wild goat, and camel) or non-ruminant herbivores (horse, donkey, and elephants) (Table 1). Among 35 identified species, 19 species were isolated from rumen contents and 16 species were isolated from fecal samples. During the 1980s, three filamentous type fungi including Neocallimastix, Orpinomyces and Piromyces and one bulbous type fungus, Caecomyces, with a total of 5 species were isolated and described from New Zealand (1), Canada (2) and United Kingdom (3). The new genus Anaeromyces and 3 genera with 11 species were isolated and described from Australia (1), Canada (1), United Kingdom (1), France (2), New Zealand (2) and Malaysia (4) during the 1990s and Ho and her colleagues [42] isolated 4 genera with 5 species during this period. The bulbous type fungi Caecomyces and Cyllamyces and filamentous type fungus Piromyces with a total of 3 species were isolated and described from United Kingdom (1) and Taiwan (2) during the 2000s. After the report on the use of barcode markers for identification of anaerobic fungi [15–17], Caecomyces sympodialis was the first described anaerobic fungi based on both barcode marker and morphological characteristics [49]. During the last decade, 12 new genera with a total 23 species were identified due to advanced barcode marker techniques and increased needs for new enzyme resources required for bioenergy production.
| Genus | Species | Host animal | Sample | Year | Ref. |
|---|---|---|---|---|---|
| Agriosomyces | longus | Ovis orientalis | Feces | 2020 | [24] |
| Aklioshbomyces | papillarum | Odocoileus virginianus | Feces | 2020 | [24] |
| Anaeromyces | contortus | Bos taurus | Feces | 2018 | [25] |
| elegans | Bos taurus | Rumen | 1993 | [13] | |
| mucronatus | Bos taurus | Rumen | 1990 | [26] | |
| polycephalus | Babalus befullus | Rumen | 2012 | [27] | |
| robustus | Ovis aries | Feces | 2016 | [28] | |
| Buwchfawromyces | eastonii | Bulbalus bubalis | Feces | 2015 | [29] |
| Caecomyces | churrovis | Ovis aries | Feces | 2017 | [30] |
| communis | Ovis aries | Feces | 1988 | [31] | |
| equi | Equus ferus | Feces | 1988 | [31] | |
| hurleyensis | Ovis aries | Rumen | 2012 | [27] | |
| sympodialis | Bos indicus | Rumen | 2007 | [32] | |
| Capellomyces | elongates | Capra aegagrus | Feces | 2020 | [24] |
| foraminis | Capra aegagrus | Feces | 2020 | [24] | |
| Cyllamyces | aberensis | Bos taurus | Feces | 2001 | [33] |
| Feramyces | austinii | Ammotragus lervia | Rumen | 2018 | [34] |
| Ghazallomyces | constrictus | Axis axis | Feces | 2020 | [24] |
| Joblinomyces | apicalis | Capra aegagrus | Feces | 2020 | [24] |
| Khoyollomyes | ramosus | Eauus grevyi | Feces | 2020 | [24] |
| Liebetanzomyces | polymorphus | Capra aegarrus | Rumen | 2018 | [35] |
| Neocallimastix | californiae | Capra aegarrus | Feces | 2016 | [28] |
| cameroonii | Ovis aries | Feces | 2015 | [36] | |
| frontalis | Ovis aries | Rumen | 1983 | [9] | |
| hurleyensis | Ovis aries | Rumen | 1991 | [37] | |
| patriciarum | Ovis aries | Rumen | 1986 | [38] | |
| variabilis | Bos indicus | 1993 | [39] | ||
| Oontomyces | anksri | Damelus dromedarius | Rumen | 2015 | [40] |
| Orpinomyces | bovis | Bos taurus | Rumen | 1989 | [41] |
| intercalaris | Bos indicus | Rumen | 1994 | [42] | |
| joyonii | Bos taurus | Rumen | 1991 | [43] | |
| Pecoramyces | ruminantium | Bos taurus | Feces | 2017 | [44] |
| Piromyces | communis | Ovis aries | Feces | 1988 | [31] |
| cryptodigmaticus | Bos taurus | Feces | 2012 | [27] | |
| dumbonicus | Elephas maximus | Feces | 1990 | [45] | |
| finnis | Equus ferus | Feces | 2016 | [28] | |
| irregularis | Bos taurus | Rumen | 2015 | [36] | |
| mae | Equus ferus | Feces | 1990 | [45] | |
| minutus | Cervus nippon | Rumen | 1993 | [46] | |
| polycephalus | Babalus befullus | Rumen | 2002 | [47] | |
| rhizinflatus | Equus africanus | Feces | 1991 | [48] | |
| spiralis | Capra aegarrus | Rumen | 1993 | [49] | |
| Tahromyces | munnarensis | Nilgiritragus hylocrius | Feces | 2020 | [24] |
NUCLEOTIDES OF ANAEROBIC FUNGI
After the first deposition of genomic DNA sequence from Neocallimastix ssp., by Brownlee [50], 23,830 nucleotide sequences were available in NCBI database (Table 1) and the numbers were in the order of Piromyces (82.63%), Neocallimastix (9.25%), Anaeromyces (4.87%), Orpinomyces (1.65%), Cyllamyces (0.54%), Caecomyces (0.38%), Pecoramyces (0.29%), and Feramyces (0.29%). In the early 90s, the applications of cDNA library construction method using λ-ZAP II vector system were reported [51,52] and the needs of potent carbohydrate degrading enzyme for bioenergy production triggered researches on the functional genes originated from the anaerobic fungi during. Also, genome projects of JGI supported by the U.S. Department of Energy (DOE) accelerated the researches on the anaerobic fungi and resulted in large amounts of nucleotide sequences information about the anaerobic fungi during the third decade. During this period, about 17,100 out of 18,771 nucleotide sequences were acquired with transcriptome analysis method using constructed cDNA library in efforts to get the sequence information about carbohydrate degrading enzymes. Currently, whole genome sequences of Anaeromycesrobustus, Neocallimastix californiae, Pecoramyces ruminantium, Piromyces finnis and Piromyces sp. E2 are available in JGI (https://mycocosm.jgi.doe.gov/index.html). The development of NGS technologies also affected the nucleotide researches on AF during the fourth decade and total 3,865 nucleotides sequenced with NGS including Illumina system (2016), PacBio system (1833) and Roch 454 GS-FLX system (16) were deposited in NCBI DB. The number of deposited nucleotide sequences was smaller than expected because the developments of NGS technologies and bioinformatics tools made it possible to translate acquired nucleotide sequences to amino acid sequences for protein prediction.
After the first report of ITS1 and 18S rRNA sequences (AF170188.10 [17], 1,160 nucleotide sequences including 210 partial sequences from Anaeromyces were deposited in NCBI DB and most of them (925) were acquired with whole genome sequence analyses. The sequencing methods were PacBio sequencing system (925), Sanger method (115) and Illumina sequencing system (19) and. The number of reported ribosomal RNA sequences from Anaeromyces was 194 including ITS1 (186), 5.8S rRNA (149) and 28S rRNA (68) sequences and those were used for identification of anaerobic fungi. Only 16 mRNA sequences including carbohydrate hydrolyzing enzymes, polysaccharide lyase (PL), esterases, phosphatase, and acyltrasferase were reported. The carbohydrate hydrolyzing enzyme sequences including cellulose (cel 1, GI 33327793), lichenase (licB, GI 33327791), glycosyl hydrolase family (GH) 1 (MH043785.1), GH10 (MH043796.1), GH32 (MH043815.1), and GH 67 (MH043829.1) were reported. In addition, three carboxyl esterases (CE) (CE7; MH043852.1, CE 12; MH043857.1, MH043856.1) and two polysaccharide lyases (PL) (PL9; MH043867.1, PL11; MH043868.1) sequences were also reported.
After the first report about 18S ribosomal RNA (M62707.1) originated from Caecomyces communis in 1993, 91 nucleotide sequences including 77 partial sequences were available in NCBI DB and the sequences were acquired with Sanger method (45) and Illumina system (23). Most of the nucleotides (68) from Caecomyces were related to fungal identification including ITS1 (61), 5.8S rRNA (61), 18S rRNA (22) and 28S rRNA (9) and only 20 nucleotide sequences were related to the information about the fungal protein. The sequences related to glycosyl hydrolase families (GH3; MH043789.1, GH8; MH043793.1, GH11; MH043803.1, GH18; MH043803.1, GH43; MH043822.1, and GH115; MH043838.1), carboxyl esterases (CE2; MH043845.1, CE3; MH043846.1, and CE15; MH043860.1) and polysaccharide lyase (PL9; MH043865.1) from Caecomyces were reported. One interesting sequence was the immunity protein 51 which was not reported from other anaerobic fungi but bacteria as a hypothetical protein.
The number of partial nucleotide sequences originated from genus Cyllamyces in NCBI DB was 122 out of total 130 nucleotide sequences and 113 nucleotide sequences were from uncultured Cyllamyces. The sequences related to ribosomal RNA were 122 including ITS1 (64), 5.8S rRNA (64), 18S rRNA (118), and 28S rRNA (4) and 8 sequences were related to the patent of xylose isomerase production. However, no mRNA sequences related to protein were reported yet.
The number of partial nucleotide sequences originated from genus Feramyces in NCBI DB was 65 out of total 69 nucleotide sequences and the sequences were acquired with Sanger method (46) and Illumina system (23). The number of ribosomal RNA sequences were 44 including ITS1 (28), 5.8S rRNA (25) and 28S rRNA (16) sequences. The nucleotide sequences related to carbohydrate degrading proteins including glycosyl hydrolase (GH2; MH043788.1, GH5; MH043790.1, GH10; MH0437951, GH30; MH043812.1, GH43; MH043823.1, GH47; MH043824.1, GH48; MH043825.1, GH76; MH043830.1, MH043831.1 GH97; MH043835.1, and GH130; MH043840.1), carboxyl esterase (CE1; MH043842.1, CE2; MH043843.1, CE4; MH043847.1, and CE12; MH043854.1), α-amylase (MH044722.1) and galactoside-O-acetyltransferase (MH043885.1) were deposited by Murphy and coworkers (http://ncbi.nlm.nih.gov/nuccore).
After the first report on AT-rich region of Neocallimastix LM-2 DNA (X14665.1) [50], 2,111 nucleotide sequences including 1,152 mRNAs and 163 rRNAs including ITS1 (140), 5.8S (110), 18S (98) and 28S (35) have been deposited in NCBI DB. In addition, 45 nucleotides were patent sequences related with AXEs, xylanases or enzyme production. The genes related with 11 glycosyl hydrolases (GH1, GH16, GH17, GH19, GH28, GH32, GH35, GH36, GH43, Gh64, and GH108), 4 carboxyl esterases (CE2, CE4, CE13, and CE16), glucanases (CelA, CelB, CelD, and Cel48) and cellobiohydrolases (CBH6, CBH20). Most nucleotide sequences were acquired with either ZAP II cDNA libraries (997) during transcriptome analyses or shotgun assembly (713) during whole genome analyses and used sequencing technologies were PacBio sequencing system (705), Sanger dideoxy sequencing method (78) or Illumina system (26).
The evolutionary closeness of cyclophilin originated from Orpinomyces sp. PC-2 and human were the first report of the nucleotide information originated from the genus Orpinomyces [53] and total 394 nucleotide sequences including 195 rRNAs and 55 mRNAs in NCBI DB. In addition, 12 nucleotide sequences were related to information about patent and 47 ITS1, 115 5.8S, 141 18S, and 77 28S rRNA sequences were related to fungal classification. The nucleotide sequences of 4 cellulases, 9 cellobiohydrolases, 8 glycosyl hydrolases, 2 glucanohydrolases, beta-glucosidase and endo-glucanase were deposited. Besides, the nucleotide sequences of other 3 hexose-hydrolases including α-amylase, lichenase, mannase and 3 pentose-hydrolases including xylanase and xylose isomerase were deposited in NCBI DB.
The whole genome sequences of Orpinomyces sp. strain C1A was reported by Yousseff and her colleagues in 2013 [54] and Orpinomyces sp. strain C1A was reclassified as Pecoramyces ruminatium strain C1A in 2017 [44]. Among available 70 nucleotide sequences, 32 mRNAs sequences and 21 rRNA sequences including ITS1 (15), 5.8S (7), 18S (3), and 28S (6) rRNA sequences originated from the genus Pecoramyces were deposited in NCBI DB. In addition, 16 nucleotide sequences related with carbohydrate degradation including α-amylase, β-glucosidase (BGL1 and BGL3), cellulase (Cel6A, Cel6B, and Cel48), glycosyl hydrolase (GH18, GH31, GH39, GH53, GH78, and GH88), polysaccharide lyase (PL1), and xylanase (XYL11 and xylan 1,4-β-xylosidase) originated from Orpinomyces were available.
Most of Piromyces nucleotide sequences were acquired from expressed sequence tag (EST) sequences and 9 nucleotide sequences were from genome sequences including shotgun sequences, and chromosome sequences. Interestingly, 258 nucleotide sequences were from patents and 94 nucleotide sequences were ribosomal RNA sequences. In Piromyces, 10 glycosyl hydrolases (GH1, GH5, GH6, GH8, GH25, GH26, GH31, GH43, GH57, and GH 127), 3 carboxyl esterases (CE4, CE12 and CE15), 12 glucanases (Cel1B, Cel1C, Cel1D, Cel3A, Cel5, Cel6A, Cel6B, Cel6C, Cel6E, Cel6G, Cel9, and Cel48), 7 cellobiohydrolases (CBHB, CBH6, CBH29, CBH120, CBHYW23-1, CBHYW23-2, and CBHYW23-4), 2 polysaccharide lyase (PL4 and PL9), xylanases and xylose isomerases were reported.
Only a limited number (12) of nucleotide sequences from genus Buwchfawromyces were available on NCBI DB and no information about functional genes from Buwchfawromyces has been reported yet. In addition, the numbers of nucleotide sequence from genus Liebetanzomyces and genus Oontomyces were 4 and 3, respectively, and all of them were ribosomal RNAs acquired with Sanger dideoxy sequencing method.
PROTEINS OF ANAEROBIC FUNGI
Until the end of the third decade, the total number of deposited protein amino acid sequences in NCBI DB was 189 even with the application of EST methods (Table 2). The length of sequence acquired from EST was ranged approximately from 500 to 800 nucleotides and this was not long enough to predict protein under the limited number of protein information. In addition, other working horse steps such as rapid amplification of cDNA ends (RACE) PCR method should be performed to get a complete amino acid sequence of the target protein [55]. It could be easy to get complete sequence information of expressed proteins with λ-ZAP II vector system, however either enough protein database or proper substrates for enzyme reactions to get the functional information about expressed proteins. The development of NGS technologies and their application to cDNA library of AF resulted in remarkable increase in sequence information of fungal proteome during the last two decades (Table 2). The number of sequence information acquired with PacBio system was 44,026 protein sequences from thee genera including Neocallimastix (20,214 proteins), Anaeromyces (12,833 proteins) and Piromyces (10,979 proteins) and that with Illumina system was 14,618 proteins from three genera including Piromyces (14,613 proteins), Neocallimastix (4 proteins) and Orpinomyces (1 proteins). On the other hand, the classical Sanger dideoxy sequencing method was also used for sequence analyses of 14,618 proteins.
Protein separation using two dimensional gel electrophoresis (2D-GE), sequence analysis using mass spectrometry (MS) and protein identification are three key steps for classic proteome analysis technique and this could be a quite powerful technique to collect and compare each protein information (Fig. 2) [56]. However, it requires long training time to get high quality results of 2D-GE due to lots of tricky steps and the repeatability of 2D-GE is comparatively low. Technical advances in MS, protein separation and sequence analyses could be performed with a single run of LC-MS and this method was regarded as more effective one than 2D-GE.
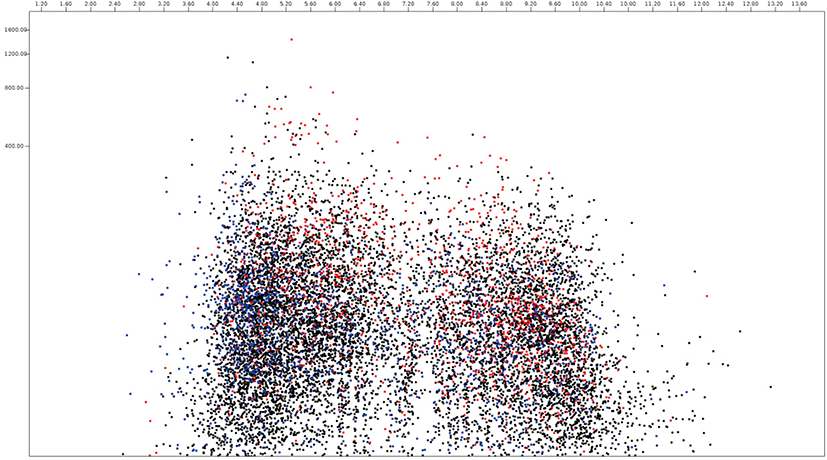
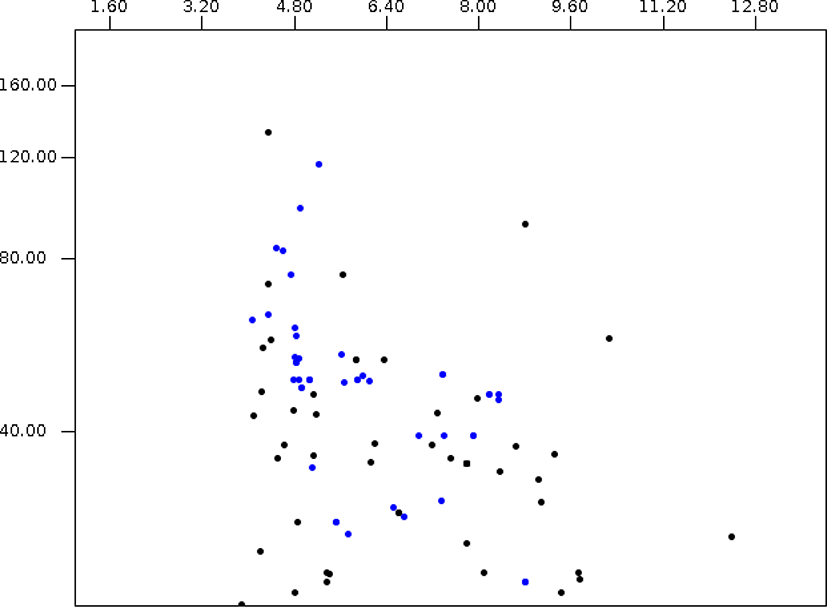
In this paper, the protein virtual gels of individual fungus based on publically available amino acid sequences were constructed using JVirGel version 2.0 software (jvirgel.de). Among 12,853 protein sequences from Anaeromyces, 1,383 and 2,874 proteins were predicted as secretory proteins and membrane proteins and 5,452 proteins were predicted as remaining proteins that were not predicted either of them (Fig. 3, Supplementary Table S1). For the total proteome of Anaeromyces, the pI values were ranged from 3.00 (hypothetical protein, ORX83422.1) to 11.10 (hypothetical protein, ORX75296.1) and molecular weights were ranged from 200 kDa (Ketoacyl-synt-domain-containing protein, ORX78811.1) to 12.7 kDa (hypothetical protein, ORX75296.1). Among known functional proteomes, the domain of unknown function (DUF) 6 containing protein (ORX84229.1) with pI 9.82 had the highest pI and cellulose-domain containing protein (ORX78609.1) with pI 3.80 had the lowest pI among functionally known secretomes. The abi-domain containing protein (ORX55221.1) with 15 kDa was the smallest and P-loop containing nucleoside triphosphate hydrolase protein (ORX86651.1) with 182.4 kDa was the largest among functionally known secretome. The average pI and molecular weight of Anaeromyces secretome were 5.88 and 55.4 kDa, respectively. The MFS (major facilitator superfamily) transporter (ORX87586.1) with pI 9.99 had the highest pI and WD40 (β– transducin) repeat-like protein (ORX61822.1) with pI 3.88 had the lowest pI among functionally known membrane proteome of Anaeromyces. The keotacyl-synthetase domain containing protein (ORX78811.1) with 199.8 kDa was the largest and RER1 protein (ORX55221.1) which involved in the retrieval of endoplasmic reticulum membrane protein with 16 kDa was the smallest among functionally known predicted membrane proteome. The average pI and molecular weight of membrane proteome were 6.88 and 69.4 kDa, respectively.
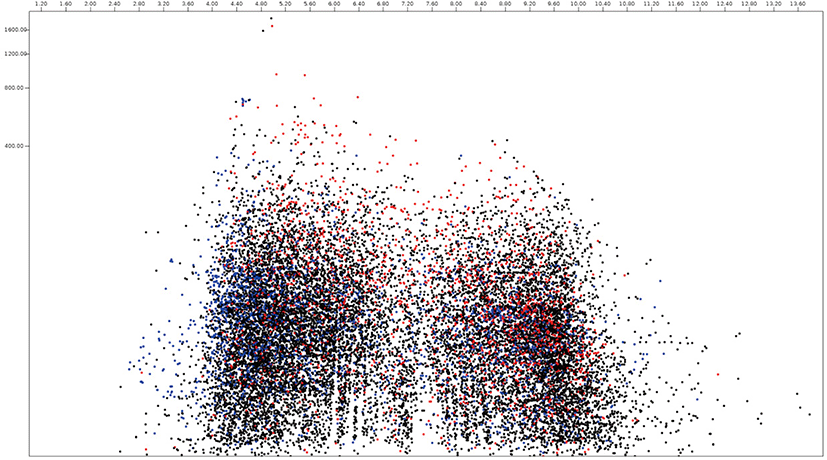
Among 15,745 protein sequences from Neocallimastix, 2,095 and 4,335 protein sequences were predicted as secretome and membrane proteome, respectively, and 9,315 protein sequences were predicted as remaining proteome (Fig. 4, Supplementary Table S2). The molecular weight of Neocallimastix proteome was a range from 200 kDa (hypothetical protein LY90DRAFT_666922, ORY72815.1) to 12.1 kDa (partial sequence of hypothetical protein LY90DRAFT_664663 from Neocallimastix californiae ORY79598.1) with an average of 58.8 kDa and hypothetical protein LY90DRAFT_499033 (ORY85919.1) with 13.8 kDa was the smallest among complete proteomes. One of hypothetical protein (ORY54617.1) with pI 10.0 had the highest pI, and another hypothetical protein (ORY24039.1) with pI 2.97 had the lowest pI among predicted secretomes of Neocallimastix. However, RNI-like protein (ORY54484.1) which involve in protein binding with pI 9.95 had the highest pI and invertase (ORY74006.1) with pI 3.84 had the lowest pI among functionally known secretomes of Neocallimastix. Scaffolding (ORY55229.1) which involved signaling pathway with 196 kDa was the largest and MFS general substrate transporter (ORY80567.1) with 14.3 kDa the smallest functionally known predicted secretome of Neocallimastix. The average pI and molecular weight of Neocallimastix secretome were 6.06 and 53.6 kDa, respectively. The pI 9.99 of essential protein for acyl-CoA-dependent ceramide synthesis, LAG1-domain containing protein (ORY33646.1) was the highest and the pI 3.88 of glycoside hydrolase/deacetylase (ORY79242.1) was the lowest among functionally known predicted membrane proteome of Neocallimastix. The protein related to the retrieval of early ER protein, Rer1 (ORT22199.1) with 15.7 kDa was the smallest and P-loop containing nucleoside triphosphate hydrolase protein (ORY43884.1) was the largest among functionally known predicted membrane proteomes of Neocallimastix. The average pI and molecular weight of Neocallimastix membrane proteome were 6.96 and 66.3 kDa, respectively.
Among 126 Orpinomyces proteomes, 29 protein sequences were predicted as membrane proteome and 97 protein sequences were predicted as remaining proteins (Fig. 5, Supplementary Table S3). The molecular weight of reported Orpinomyces proteome was ranged from 133.1 kDa (partial sequence of hypothetical protein from Orpinomyces sp. OUS1, CAI29548.1) to 10.1 kDa (partial sequence of the hypothetical protein from Orpinomyces sp. OUS1, CAI29549.1) with average 38.7 kDa. In addition, the pI of reported Orpinomyces proteome was ranged from 12.35 (partial sequence of the hypothetical protein from Orpinomyces sp OUS1, CAI29565.1) to 3.23 (partial sequence of the hypothetical protein from Orpinomyces sp OUS1, CAI29558.1) with an average of 6.47. The GH 95 from Orpinomyces joyonii (AWI66988.1) with 117.2 kDa was the largest and Cyclophilin B precursor from Orpinomyces joyonii (ABC47329.1) with 22.0 kDa was the smallest Orpinomyces membrane protein. In addition, pI value of reported Orpinomyces membrane proteins was ranged from 9.29 (GH114 from Orpinomyces joyonii, AWI66991.1) to 4.08 (partial sequence of putative 5-azacytidine resistance protein from Orpinomyces sp. OUS1, CAI11365.1).
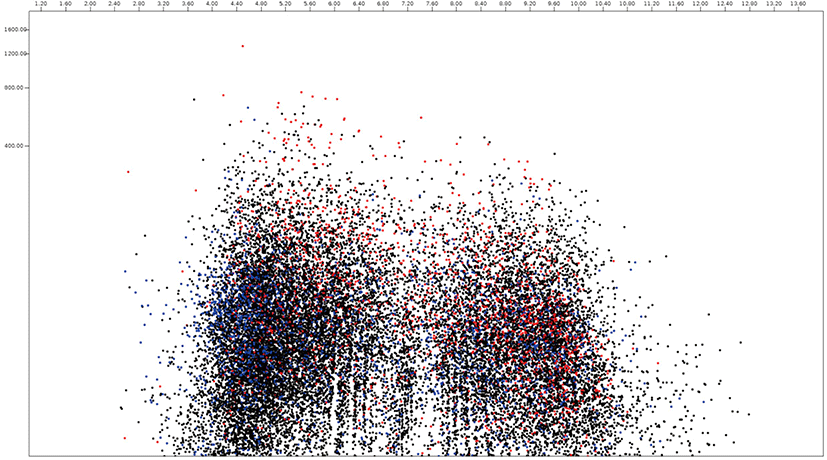
Among 18,822 Piromyces proteomes, 2,270 and 4,863 protein sequences were predicted as secretome and membrane proteome, respectively, and 11,689 protein sequences were predicted as remaining proteins (Fig. 6, Supplementary Table S4). The molecular weight of Piromyces proteome was ranged from 200.0 kDa (Hypothetical protein from Piromyces sp. E2, OUM64425.1) to 12.9 kDa (hypothetical protein from Piromyces sp. E2, OUM60062.1) with an average of 56.9 kDa and the pIs were ranged from 11.00 (hypothetical protein from Piromyces finnis, ORX60159.1) to 2.92 (hypothetical protein from Piromyces sp. E2, OUM67877.1) with average of 6.57. The pI 10.06 of DUF6-domain-containing protein (ORX43281.1) was the highest and the pI 2.95 of non-catalytic module family DOC2 (OUM64499.1) was the lowest among functionally known predicted secretomes of Piromyces. The enzyme endoglucanase 5A with 192.9 kDa was the largest and putative terbinafine resistance locus protein (ORX49356.1) with 13.1 kDa was the smallest among functionally known predicted secretomes of Piromyces. The average pI and molecular weight of Piromyces secretome were 6.03 and 49.9 kDa, respectively. The glycosyltranferase family 2 protein (OUM61397.1) with 193.3 kDa was the largest and putative terbinafine resistance locus protein (ORX49356.1) with 13.1 kDa was the smallest among functionally known predicted membrane proteomes of Piromyces.
CARBOHYDRATE DEGRADING PROTEINS
The information about the proteins related to carbohydrate degradation including glycoside hydrolase (GH), glycosyl transferase (GT), polysaccharide lyase (PL), carbohydrate esterase (CE) and carbohydrate binding module (CBM) as an associated module is publically available on the CAZY DB (www.cazy.org). The GH family proteins hydrolyze the glycosidic bonds between sugar or bond between sugar and non-sugar moiety and 165 GH families have been registered to CAZY database. From anaerobic fungi, 36 GH family were registered and 5 different GH clans including (α/α)6, (β/α)8, 5-fold β-propeller and β-jelly roll were reported. The hexose hydrolyzing enzymes including α-glucosidase (EC 3.2.1.20; GH31), β-glucosidase (EC 3.2.1.21, GH1 and GH3) which hydrolyze non-reducing glucosyl residue terminal were detected from Caecomyces, Neocallimastix, Orpinomyces, Pecoramyces and Piromyces. The enzyme endoglucanases (EC 3.2.1.4; GH5, GH6, GH9, GH45, and GH 48) which hydrolyze glucosidic linkage of cellulose, lichenin and glucans were detected from Anaeromyces, Neocallimastix, Orpinomyces and Piromyces. The enzyme α-amylases (EC 3.2.1.1; GH13 and GH57) which hydrolyze α-D-glycosidic linkages of polysaccharide were detected from Orpinomyces, Pecoramyces and Piromyces. The enzyme β-mannase (EC 3.2.1.78; GH26) is a representative enzyme of GH 26 family proteins and the glucanase (EC 3.2.1.39) is the representative enzyme of GH 17 and GH 64 family proteins. The enzyme α-galactosidase (EC 3.2.1.22) and β-galactosidase (EC 3.2.1.23) were the representative enzymes of GH 36 and GH 35 family proteins, respectively. The enzymes involved in the hydrolyses of pentose sugar including endo-1,4-β-xylanase (EC 3.2.1.8, GH10 and GH11), β-xylosidase (EC 3.2.1.37, GH43 and GH 120), α-L-fucosidase (EC 3.2.1.51, GH 95 and GH 141), α-L-rhamnosidase (EC 3.2.1.40, GH 28 and GH 78), β-L-arabinofuranosidase (EC 3.2.1.185, GH 127) were detected from anaerobic fungi. In addition, chitinase (EC 3.2.1.14; GH18 and GH19) and chitosanse (EC 3.2.1.132; GH8) were also detected from anaerobic fungi.
The glycosyl transferases transfer sugar during glycosidic bond synthesis and 5 glycosyl transferase were registered and 3 polysaccharide lyase (PL), pectate lyase (EC 4.2.2.2, PL1), rhamnogalacturona endolyase (EC4.2.2.23, PL4) and rhamnogalacturona exolyase (EC 4.2.2.24, PL11) were detected from anaerobic fungi. The carbohydrate esterases (CE) hydrolyze esters into an acid and an alcohol and 8 CE families were detected from anaerobic fungi. The enzyme acetyl xylan esterase (EC 3.1.1.72), feruloyl esterase (EC3.1.1.73), cinnamoyl esterase (EC 3.1.1-) and carboxylesterase belong to CE 1 family, the existence of only acetyl xylan esterase and feruloyl esterase were reported. The acetyl xylan esterase from Neocallimastix frontalis PMA02 cleaves ester bond between acetyl side group and xylan or xylo-oligosaccharides [57] and feruloyl esterase from Anaeromyces mucronatus cleaves ester bond between ferulate and polysaccharide [58]. The representative enzyme of CE 8 is pectin methylesterase (EC 3.1.1.11) and the partial sequence of CE 8 from Orpinomyces joyonii D3B (AWI67007.1) could be methylesterase. Likewise, partial sequence of CE 13 from Neocallimastix cameroonii G3 (AWI67012.1) and CE 15 from Piromyces sp could be pectin aceylesterase (EC 3.1.1.-) and 4-o-methyl-glucuronoyl methylesterase (EC3.1.1.-), respectively. The protein 3D structure of CE 1, CE3, CE6, and CE13 family were (α/β/α)-sandwich type and those of CE 4, CE 8 family were (β/α)7-barrel and (β)-helix type, respectively. The carbohydrate esterases do not directly hydrolyze glucosidic linkage of polysaccharides, however, glucose hydrolysis is promoted with the removal of non-sugar side groups by CE [57].
RESEARCHES ON ANAEROBIC FUNGI
The research articles about anaerobic fungi were searched on PubMed (PM) and PubMed Central (PMC) DB using individual genus name as keywords and the numbers of hits were 1,138 in PM and 1,344 in PMC during the period from 1980 to 2019 (Table 3). Before 1980, 5 journal articles were detected in PM and one was detected in PMC. After filtration by deleting duplicated one, the numbers were reduced to 444 in PM and 719 in PMC and the articles overlapped both PM and PMC were 132 (Supplementary Table S5). Most of the results acquired from PM were directly related to anaerobic fungi, however, some results from PMC were not directly related to keyword or unrelevant to it. According to PM results, Neocallimastix had been the most popular topic in the anaerobic fungal research until 1990 because it was the earliest genus of anaerobic fungi. In addition, the numbers of research articles about Neocallimastix were the highest both in PM and PMC database and those about Orpinomyces and Piromyces were the next. The researches on Piromyces were continuously reported last four decades. Even with their early discovery, researches on Anaeromyces, and Caecomyces were actively reported during the 2000s. The research on newly discovered anaerobic fungi including Buwchfawromyces, Feramyces, Liebetanzomyces, Oontomyces, and Pecoramyces were reported during 2010s.
The AF does not possess mitochondria but hydrogenosome for ATP production [59] and the hydrogenosome is surrounded by the double membrane [60]. The hydrogen can be produced through enzyme cascade reactions in hydrogenosome [61] and the biochemical characteristics of hydrogenosomal enzymes including pyruvate format-lyase [62], hydrogenase [63] and malic enzyme [64] were reported.
The first report on the physicochemical mechanism of AF in fiber degradation [3] suggested the importance of AF during fiber digestion in the rumen and it triggered the researches on the possible use of AF themselves or their enzymes as feed additives to increase the fiber digestibility in the rumen. The glycosidase activities in the culture supernatant of Neocallimastix frontalis (N. frontalis) using filter paper or avicel as substrate were compared [65] and the substrate conditions were expanded to natural fiber including wheat straw [6], Italian ryegrass [66] and maize stem [67]. The β-glucosidase [68] and xylanases [69] from N. frontalis were purified and characterized through the biochemical procedure using chromatograms and cellulases (celA, celB, and celC) from Neocallimastix patriciarum (N. patriciarum) were produced through molecular procedure using cDNA cloning and consecutive heterologous expression in Escherichia coli [70]. Later, xylanase and mannanase from Piromyces [51], cellulase and xylanases from Orpinomyces [71], xylanases from Anaeromyces [72] and cellulase from Pecoramyces [73] were reported.
Unlike other eukaryotic genes, no introns were detected from most of endoglucanases [74,75] originated from AF and the horizontal transfer of bacterial genes to AF could be the reason for the intronless endoglucanases in AF [75,76]. The horizontal gene transfer in AF is crucial for the survival of AF in the rumen [77] and it could be beneficial to researchers for the production of AF protein. The use of cDNA library constructed with vector system was considered a promising tool for the production of enzymes from AF and has been used for three decades. The development NGS system with bioinformatics enormously increased the amount of protein sequence information with a single run and assembled 27,560 transcripts were acquired with 70.2 Gb reading with Illumina HiSeq 2,000 platform [78]. The Illumina systems cover their short read length (up to 300 bp) with a huge amount of read (up to 6,000 Gb for NovaSeq), however, there might be some possibilities for misleading due to the characteristics of anaerobic fungal proteins. Recently, Pacific Biosciences developed PacBio system which armed with long read length (15–20 kb), however solid reports have not been published yet.
Natural celluloses were classified as cellulose I, II, III, and IV based upon its crystalline allomorphs [79] and the possible effects of cellulose crystallinity index on cellulose degradability was proposed [80]. The adsorption and activity of cellulolytic enzymes from AF were affected by the cellulose microcrystallinity [81] and the protein, cellulose binding module (CBM) in cellulases could be responsible for ligand binding action [82]. After molecular and biochemical characterization of CBM29 from Piromyces equi [83], 11 CBM including CBM1, 6, 10, 13, 18, 22, 26, 29, 35, 52, and 66 from Anaeromyces, Caecomyces, Neocallimastix, Orpinomyces, Pecoramyces and Piromyces are available in CAZY database (www.cazy.org). Anaerobic fungi improved their survivability under various substrate conditions with a wide variety of carbohydrate degrading enzymes which consisted of combinations of GH and CBM domains and this could be the reason why AFs maintain a wide range of enzyme systems for fiber digestion. The use of enzyme cocktail has been used in the glycosylation process of fibrous biomass for biofuel production and the similar concept was proposed using enzyme cocktail form AF [71]. The research on designing chimeric enzymes using AF genes in efforts to make multipurpose with thermostability was reported [84].
SUMMARY
Anaerobic fungi produce a wide variety of powerful carbohydrate hydrolyzing enzymes that can be used for animal production, biofuel production, food production and other purposes. The difficulties in the cultivation of AF under strictly anaerobic conditions caused hesitation in AF researches, however, the social requirement of substitutional energy source to petroleum and the development of molecular techniques including NGS stimulated researches on AF. During the last decades, development in culture medium led the discovery of new 12 genera of AF, however, there might be possibilities for more hidden AF in untouched area. In addition, large amounts of protein sequences have been produced during the last two decades, however, the information was skewed to three genera including Piromyces, Neocallimastix and Anaeromyces. The researches on AF should be expanded not only to newly described genera but the target itself. With a massive amount of sequence data, it might be difficult to scrutinize each protein, and researchers normally screen target sequences mechanically. However, sometimes AFs bear unexpected gift which researchers can easily miss.