RESEARCH ARTICLE
Complete genome sequence of Salmonella enterica strain K_SA184, multidrug resistance bacterium isolated from lamb (Ovis aries)
Hyeri Kim1,#
,
Jae Hyoung Cho1,#,
Jin Ho Cho2,#,
Minho Song3,#,
Hakdong Shin4,
Sheena Kim1,
Eun Sol Kim1,
Hyeun Bum Kim1,*,
Ju-Hoon Lee5,*
Author Information & Copyright ▼
1Department of Animal Resources Science, Dankook University, Cheonan 31116, Korea
2Division of Food and Animal Science, Chungbuk National University, Cheongju 28644, Korea
3Division of Animal and Dairy Science, Chungnam National University, Daejeon 34134, Korea
4Department of Food Science and Biotechnology, College of Life Science, Sejong University, Seoul 05006, Korea
5Department of Food and Animal Biotechnology, Department of Agricultural Biotechnology, Center for Food and Bioconvergence, Seoul National University, Seoul 08826, Korea
*Corresponding author: Hyeun Bum Kim, Department of Animal Resources Science, Dankook University, Cheonan 31116, Korea. Tel: +82-41-550-3653, E-mail:
hbkim@dankook.ac.kr
*Corresponding author: Ju-Hoon Lee, Department of Food and Animal Biotechnology, Department of Agricultural Biotechnology, Center for Food and Bioconvergence, Seoul National University, Seoul 08826, Korea, Tel: +82-2-880-4854, E-mail:
juhlee@snu.ac.kr
#These authors contributed equally to this work.
© Copyright 2021 Korean Society of Animal Science and Technology. This is an Open-Access article distributed under the terms of the
Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits
unrestricted non-commercial use, distribution, and reproduction in any
medium, provided the original work is properly cited.
Received: Sep 10, 2020; Revised: Oct 22, 2020; Accepted: Oct 23, 2020
Published Online: Jan 31, 2021
Abstract
Salmonella enterica is a representative foodborne pathogen in the world. The S. enterica strain K_SA184 was isolated from the lamb (Ovis aries), which was collected from a local traditional market in South Korea. In this study, the S. enterica strain K_SA184 was sequenced using PacBio RS II and Illumina NextSeq 500 platforms. The final complete genome of the S. enterica strain K_SA184 consist of one circular chromosome (4,725,087 bp) with 52.3% of guanine + cytosine (G + C) content, 4,363 of coding sequence (CDS), 85 of tRNA, and 22 of rRNA genes. The S. enterica strain K_SA184 genome includes encoding virulence genes, such as Type III secretion systems and multidrug resistance related genes.
Keywords: Salmonella enterica K_SA184; Lamb (Ovis aries); Whole genome sequencing; Antimicrobial resistance; Type III secretion systems
INTRODUCTION
Salmonella is a representative foodborne pathogen which is the most commonly identified in poultry, eggs and dairy products. The most common symptom of salmonella infection is gastroenteritis, follow by bacteremia and enteric fever. Most forms of poultry meat, pork, and beef are the main sources responsible for salmonella infection [1] because the contamination of the organ and carcass with salmonella easily occurs during the slaughtering process of the food animals at abattoirs [2].
The Salmonella enterica strain K_SA184 was isolated from a lamb (Ovis aries) purchased from the local traditional market in Suwon, Gyeonggi-do, Korea. The S.enterica strain K_SA184 was streaked to xylose lysine tergitol 4 (XLT4) agar and incubated at 37°C for 24 h. The suspected colony in XLT4 agar was inoculated into Luria-Bertani (LB) broth and incubated at 37°C for 24 h. To analyze the complete genome, the S. enterica strain K_SA184 was sequenced by PacBio RS II (Pacific Biosciences, Menlo Park, CA, USA) at Insilicogen (Yong-in, Korea) and Illumina NextSeq 500 (Illumina, San Diego, CA, USA) platform at LabGenomics (Seongnam, Korea) [3]. The genomic DNA of S. enterica K_SA184 for PacBio and Illumina sequencing were extracted using the MagAttract HMW DNA Kit (QIAGEN), and NucleoSpin® Microbial DNA kit (TAKARA) according to the manufacturer’s instructions. Library preparation was conducted using SMRTbell™ Template Prep Kit 1.0 for Pacbio (Pacific Biosciences) and TruSeq DNA Sample Preparation Kit for Illumina (Illumina) according to the manufacturer’s instructions. PacBio sequencing yielded 1,474,738,487 base pairs and 190,304 long reads after filtering, and 5,513,948 paired-end reads with 832,606,148 bp was obtained with Illumina sequencing. De novo assemble was conducted using the hierarchical genome assembly process (HGAP v2.3.0) workflow and polished using Quiver. Subsequently, Illumina NextSeq reads were aligned to the PacBio RSII assembly using Burrows-Wheeler Aligner (BWA)-MEM v0.7.17-r1188, and the errors were corrected by using Pilon version 1.23 [4,5]. The quality of genome assembly and the validation of the final genome were assessed by using Quality Assessment Tool for Genome Assemblies (QUAST) v5.0.2 and Benchmarking Universal Single-Copy Orthologs (BUSCO) v3.0.2 [6,7].
Open reading frames (ORFs) and RNA genes of S. enterica strain K_SA184 were predicted and functionally annotated by rapid prokaryotic genome annotation (PROKKA) v1.14.5 and Rapid Annotation using Subsystem Technology (RAST) v2.0. The functional categorization and classification of all predicted ORFs were conducted using the RAST server-based SEED viewer and Clusters of Orthologous Groups (COG) – based EggNOG. The putative virulence factors and Antimicrobial resistance were described using BLAST according to the Virulence Factor Database (VFDB) and antibiotic resistome surveillance with the comprehensive antibiotic resistance (CARD) [8,9]. The whole genome of S. enterica strain K_SA184 is composed of one circular chromosome (4,725,087 bp) with 52.3% of guanine + cytosine (G + C) content, 4,363 of coding sequence (CDS), 85 of tRNA, and 22 of rRNA genes.
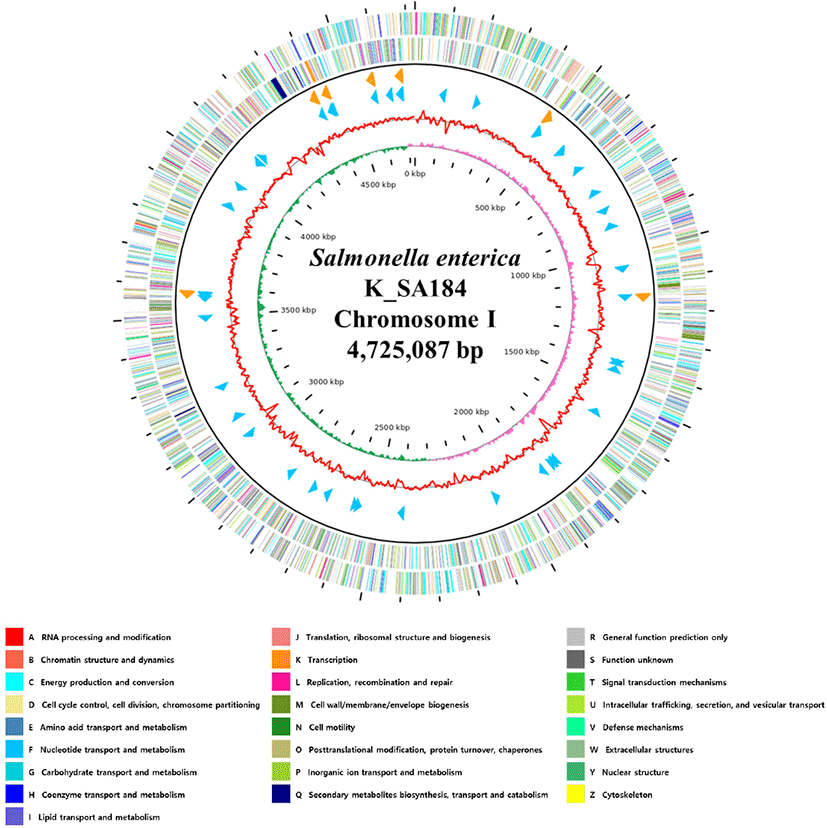
The complete genome of the S. enterica strain K_SA184 contains the virulence genes encoding Salmonella pathogenicity island 1 & 2 Type III secretion systems which serve several pathogenic functions in killing of macrophages and in interference with immune responses as reported by others [10]. Furthermore, the S. enterica strain K_SA184 also possesses multidrug resistance coding genes which are associated with a variety of drugs resistance Efflux Pumps (mdtk) and Resistance to fluoroquinolones, such as, cephalosporins (AmpC), and fluoroquinolones (Par, Gyr). We summarized the general properties of the S. enterica strain K_SA184’s complete genome in Fig. 1 and Table 1. The further in-vivo studies using S. enterica strain K_SA184 will help us to decipher the potential roles of the virulence genes in the pathogenesis.
Fig. 1.
Genome map of Salmonella enterica strain K_SA184.
The outer circle denotes the locations of all annotated open reading frames (ORFs), and the inner circle with the red denotes guanine + cytosine (GC) content. Pink, and green peaks denote GC skew. The orange arrows denote rRNAs, and the sky blue arrows denote the tRNA operons. All annotated ORFs are colored differently based on the Clusters of Orthologous Groups (COG) assignments.
Download Original Figure
Table 1.
Genome features of Salmonlla enterica strain K_SA184
| Property |
Term |
| Libraries used |
PacBio SMRTbell™ library
TruSeq DNA Sample Preparation Kit |
| Sequencing platforms |
PacBio RS II sequencer
Illumina NextSeq 500 |
| Assemblers |
PacBio SMRT analysis v2.3.0 HGAP.3 |
| Annotation method |
PROKKA v1.14.5 and RAST v2.0 |
| Average genome coverage |
159× |
| Chromosome length (bp) |
4,725,087 bp |
| No. of contigs |
1 |
| guanine + cytosine (G + C) content (%) |
52.3 |
| Protein-coding genes (CDSs) |
4,363 |
| rRNA genes |
22 |
| tRNA genes |
85 |
| Plasmids |
0 |
| Genbank accession No. |
CP061159.1 |
Download Excel Table
Acknowledgements
We thank Mo Re Kim (Brandeis University, MA, USA) for the English grammar corrections.
Availability of data and material
Ethics approval and consent to participate
REFERENCES
Eng SK, Pusparajah P, Ab Mutalib NS, Ser HL, Chan KG, Lee LH. Salmonella: a review on pathogenesis, epidemiology and antibiotic resistance. Front Life Sci. 2015; 8:284-93


Li H. Aligning sequence reads, clone sequences and assembly contigs with
BWA-MEM. arXiv:13033997v1 [preprint] 2013[cited 2020 Aug 9]https://arxiv.org/abs/1303.3997