
INTRODUCTION
Assisted reproductive techniques (ARTs) are used to preserve the genomic source of endangered species and produce disease research models. Among existing ARTs, somatic cell nuclear transfer (SCNT) has been widely employed in agriculture and biomedical research fields in many countries and laboratories. After the first successful birth of Dolly [1], SCNT has become popular worldwide and been applied to produce many species, including cattle [2], mice [3], goats [4], pigs [5], cats [6], and rabbits [7]. However, SCNT has several limitations to its general implementation, specifically, high cost and low efficiency. Primarily, the micromanipulator required for SCNT is highly priced and difficult to handle. Therefore, the cloning efficiency depends on the competence of the experimenter.
As an alternative to the SCNT technique, a handmade cloning (HMC) technique has been developed that can clone animals without a micromanipulator [8]. The HMC method, which was established in cattle, involves removing the nucleus by bisection using a microblade [9], thus, minimal equipment and skill is required for cloning. After bisection, all halved oocytes without chromatin are reconstructed by fusing two enucleated oocytes into one cytoplast. However, this method is disadvantageous in two ways: 1) it results in mitochondria heteroplasmy of recombinant embryos and 2) half of the oocyte starting material is wasted during enucleation. In addition, the cytoplasmic lysis rate is increased during the bisection of zona-free oocytes [10]. The cytoplasmic volume of oocytes is an important factor affecting the cleavage rate and developmental competence of recombinant embryos [11], therefore, the problem of cytoplasmic loss requires a solution.
To solve this problem, a modified HMC (mHMC) method that does not involve bisection has been proposed for sheep [11,12]. The mHMC method removes a tiny fraction of cytoplasm containing the maternal chromosome using the sharp end of a Pasteur pipette under an optical microscope instead of a micromanipulator. The enucleation of mHMCs can produce cloned embryos into one oocyte by minimizing cytoplasmic loss, enabling the efficient use of experimental resources and overcoming mitochondrial heterogeneity. In practice, this method has been used to produce cloned animals including calves [13], camels [14], and goats [15]. The cytoplasm of porcine oocytes has higher lipid content than that of other animals [10], and oocytes at the MII stage are more fragile. Although pig cloning using HMC has been successfully reported [10,16,17], there are still obstacles to the widespread use of this technology.
Therefore, this study develops and evaluates a porcine mHMC method by establishing a pulled Pasteur pipette suitable for pigs, which reduces the occurrence of cytoplasmic loss and mitochondrial heterogeneity. When performing HMC and SCNT, hypertonic treatment with sucrose is used to induce the formation of protrusions around chromosomes and spindle regions in oocytes [18]. This treatment arrests chromosomes in metaphase and leads to cytoplasmic membranes containing the oocyte nucleus [19], which can help improve the enucleation efficiency [20]. Demecolcine (DEM)-assisted enucleation of oocytes has been applied to the production of bovine [21] and porcine [20,22] embryos derived from SCNT, but has not yet been applied in porcine mHMC. Therefore, this study determines the DEM treatment conditions for optimal enucleation and applies the mHMC method to pigs for the first time. Furthermore, we compare the efficiency of enucleation and in vitro developmental competence for embryos derived using SCNT and mHMC techniques.
MATERIALS AND METHODS
Chemicals for the experimental procedure were purchased from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise stated.
The donor cell line was established using porcine fetal fibroblasts (PFFs) from a 35 day-old fetus. Briefly, the fetus was recovered and minced 4–7 times with phosphate-buffered saline (PBS; Gibco, Waltham, MA, USA). Then, the head, limbs, and internal organs were removed from the fetus. The remaining tissues were chopped into small pieces and placed in PBS supplemented with 5% fetal bovine serum (FBS; Gibco) and 0.5% penicillin/streptomycin (P/S). The chopped tissues were centrifuged, resuspended, and cultured in Dulbecco’s modified eagle’s medium (DMEM; Gibco) supplemented with 10% FBS and 1% P/S at 38.5°C in a 5% CO2 and humidified atmosphere in air. When the cultured cells were confluent, they were trypsinized, washed, and stored in liquid nitrogen. For both SCNT and HMC, PFFs were thawed, cultured, and subsequently used for 4–7 passages.
Ovaries used for experimental procedures were collected from a local slaughterhouse and transported to the laboratory in physiological saline with 1% P/S sulfate at 28°C–32°C within 3 h of collection. After the ovaries arrived in the laboratory, they were washed with saline as soon as possible, cumulus-oocyte complexes (COCs) were collected from antral follicles (3–8 mm in diameter) using an 18-gauge needle with a 10-mL disposable syringe. Aspirated fluids were deposited in a 37°C water bath. The sediments were then washed twice with saline. COCs were surrounded by at least three layers of cumulus cells and a uniform cytoplasm. Selected COCs were washed in in vitro maturation (IVM) media, which consisted of tissue culture medium 199 (TCM-199; Gibco) supplemented with 2.5 mM fructose, 0.4 mM L-cysteine, 1 mM sodium pyruvate, 0.13 mM kanamycin, 10 ng/mL epidermal growth factor, 500 IU/mL gonadotropin hormone, and 10% (v/v) porcine follicular fluid. The COCs were cultured in maturation media for 22 h at 39°C in a 5% CO2 and humidified atmosphere in air. After 22 h of maturation, COCs were washed and moved into fresh IVM media, which was follicle stimulating hormone (FSH) - and human chorionic gonadotropin (hCG) -free, and further cultured for 22 h under the same conditions.
In order to determine the optimal time required to treat the DEM, COCs were cultured with 0.4 μg/mL DEM for 30 min, 60 min, 90 min, and 120 min after IVM. The DEM-treated COCs were transferred to the culture medium with 0.1% hyaluronidase dissolved in North Carolina State University (NCSU) media and cumulus cells were removed by repeated and gentle pipetting. After denuding, mature oocytes with first polar bodies and DEM-induced cytoplasmic protrusions were selected (Fig. 1).
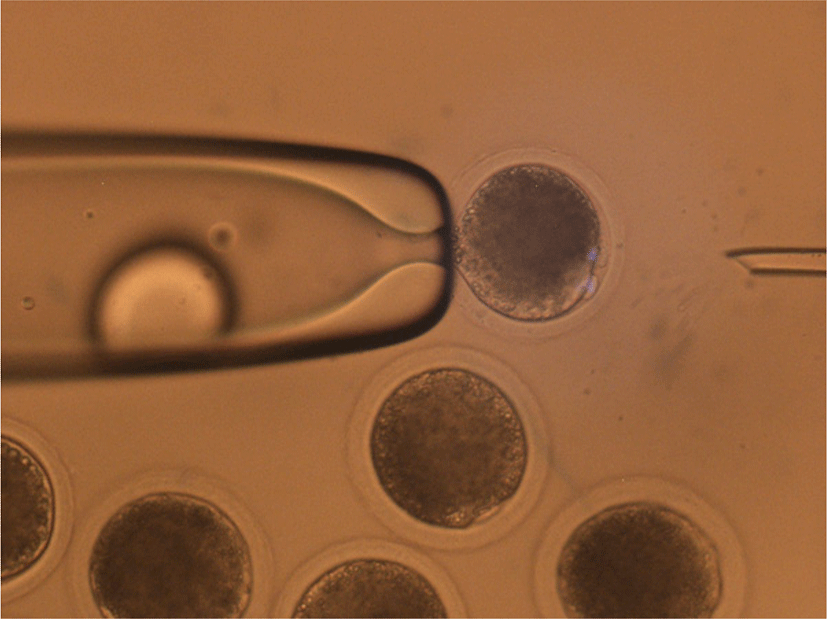
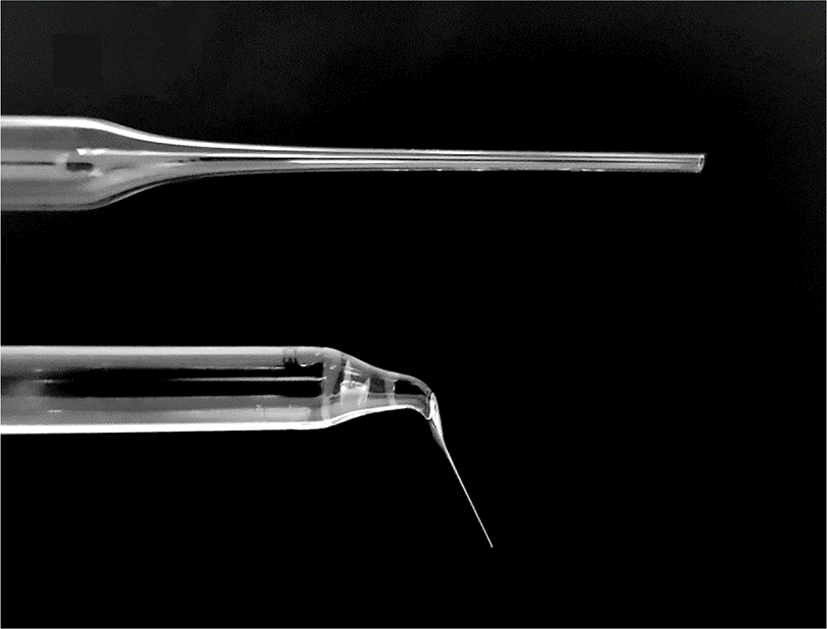
Commercial glass Pasteur pipettes were used as tools for enucleation. Briefly, after sterilizing the Pasteur pipettes, the narrow tip of the pipettes was heated on a flame by an alcohol lamp until slightly melted, then pulled rapidly until the inner diameter was close to that of the oocytes, and left for 10 s until the tip was cooled. After cooling, the tip of the pulled pipette was heated and pulled again to obtain a narrower inner diameter. Finally, the end part of the tip was cut, the cross-section of the tip was evaluated under a microscope, and aspiration pipettes were selected that had a fine and smooth tip section with a slightly larger diameter than the protrusion part of the DEM-treated oocyte cytoplasm (Fig. 2). Before use, the opposite side of the tip was filled with 1.5 mL of mineral oil to prevent capillary action.
The mature oocytes with first polar bodies and DEM-induced cytoplasmic protrusions were moved into N33 (NCSU medium with 33% FBS), then 3.3 mg/mL pronase in N33 for 30 s to partially remove the zona pellucida (ZP) that were quickly washed in N20 (NCSU medium with 20% FBS) and N2 (NCSU medium with 2% FBS) drops. Subsequently, the partially digested ZP was completely removed in N10 (NCSU medium with 10% FBS) drops using repeated pipetting. During the removal of ZP, the first polar body was also removed.
The entire process of mHMC was performed in drops covered with mineral oil. After removal of the ZP, oocytes were enucleated using aspiration pipettes. Ten to fifteen oocytes were transferred into 5 μL of N10 drop and gently rolled to find the protrusion of the MII chromosome. The tip of the aspiration pipette was attached to the surface of the protrusion area, and slight pressure was applied to the opposite side of the oocyte. When the protrusion part of the cytoplasm flowed into the inside of the aspiration pipette, the pipette was moved out from the drop while maintaining the pressure to separate the aspirated cytoplasm due to tension between the medium and mineral oil. Most of the cytoplasm remained in the drop, except for the protrusion part of the cytoplasm containing MII chromosomes. After enucleation, each oocyte was quickly transferred into N10 supplemented with cytochalasin B to prevent cytoplasmic degeneration of the oocytes.
Prior to cell attachment, 10–15 enucleated cytoplasm samples were grouped and transferred to the NCSU drop supplemented with 1 mg/mL polyhydroxyalkanoates (PHA) for 5 s. Each cytoplasm sample was then rapidly transferred into N2 drops containing fibroblasts. After attachment of cytoplasm and fibroblasts, the couplets were transferred into a fusion medium (0.28 mol/L mannitol supplemented with 0.1 mM MgSO4 and 0.05 mM CaCl2) and equilibrated.
Fusion and activation were performed in a one-step process, which was modified according to a previously described procedure. Using a cell fusion generator (LF101; NepaGene, Chiba, Japan), couplets were aligned on the wire of the BTX fusion chamber with fusion media and exposed to electrical stimulation using an alternating current (AC) of 6 V and 700 kHz. The fibroblast part of the couplet was oriented toward the end farther from the wire, after which a single direct current (DC) of 200 V/cm was fused for 9 μs. After the pulse, the couplets were detached carefully from the wire, transferred to N10 drops, and incubated for 30 s. After incubation, reconstructed embryos were washed in porcine zygote medium (PZM)-3, then incubated in 1.9 mM 6 dimethylaminopurine (6-DMAP) dissolved in PZM-3 for 3 h at 39°C in a 5% CO2 and humidified atmosphere in air. After fusion and activation, the reconstructed embryos were cultured in 400 μL PZM-3 of 4-well dish (Nunc, Roskilde, Denmark) and covered with the same amount of mineral oil as the modified Well of well (WOW) system. The WOW system was suggested for zona free embryo culture [23]. In the modified WOW system used to culture individual embryos, a small well was produced in one well of a 4-well dish by pressing a sharp steel needle heated gently by hand. Wells were filled with PBS and 5% FBS, and inner air bubbles were removed and flushed by repeated rigid pipetting. Then, PBS was replaced with PZM-3, and the wells were again covered with mineral oil. After 3 h of fusion and activation, reconstructed embryos were washed with PZM-3 and cultured in groups of 20–25 oocytes per large well at 39°C in a 5% CO2 and humidified atmosphere in air. The cleavage and blastocyst formation rates were evaluated on days 2 and 7 after culture (Fig. 3).

After treatment with DEM for 90 min, mature oocytes with extruded first polar bodies and DEM-induced cytoplasmic protrusions were placed in a microdrop of NCSU containing Hoechst 33342 and cytochalsin B. Under UV light, the first polar body and small portion of cytoplasm containing metaphase II chromosomes of oocytes were aspirated using an enucleation pipette. Enucleated oocytes were transferred to NCSU media without Hoechst 33342 and cytochalsin B, and a single donor cell with a smooth membrane was injected into the enucleated oocyte. The oocyte-cell couplets were equilibrated in 0.28 M mannitol media then placed between copper wire electrodes equipped with a micromanipulator. A single direct pulse of 200 V/mm was applied for 30 μs using a cell fusion generator. After fusion, oocytes were washed three times and kept in porcine zygote medium-3 (PZM-3) for 30 min. Thereafter, oocytes were transferred and cultured in PZM-3 containing 1.9 mM 6-DMAP. After 3 h, the oocytes with no donor cells in the perivitelline space were considered as reconstructed oocytes.
The reconstructed embryos were cultured in 40 μL of PZM-3 droplets covered by 3 mL of mineral oil in a 35-mm embryo culture dish at 38.5°C in 5% CO2 in a humidified atmosphere. The cleavage rate and blastocyst rate were determined after day 7.
To measure the levels of ROS in enucleated oocytes in each group, the ROS levels were investigated using the Image-iTTM LIVE Green Reactive Oxygen Species Detection Kit (Invitrogen, Thermo Fisher Scientific, Carlsbad, CA, USA ). In short, enucleated oocytes were transferred into Dulbecco’s Phosphate-Buffered Saline (DPBS, Gibco) containing 25 μM carboxy-H2 DCFDA, then incubated for 30 min at 39°C and in 5% CO2, protected from light. After incubation, the oocytes were washed in DPBS. The emission of fluorescence from oocytes was recorded using a camera combined with a fluorescence microscope with UV filters (460 nm for ROS). The recorded fluorescent images were analyzed using NIS-Elements BR software 3.00 (Nikon, Minato, Tokyo, Japan).
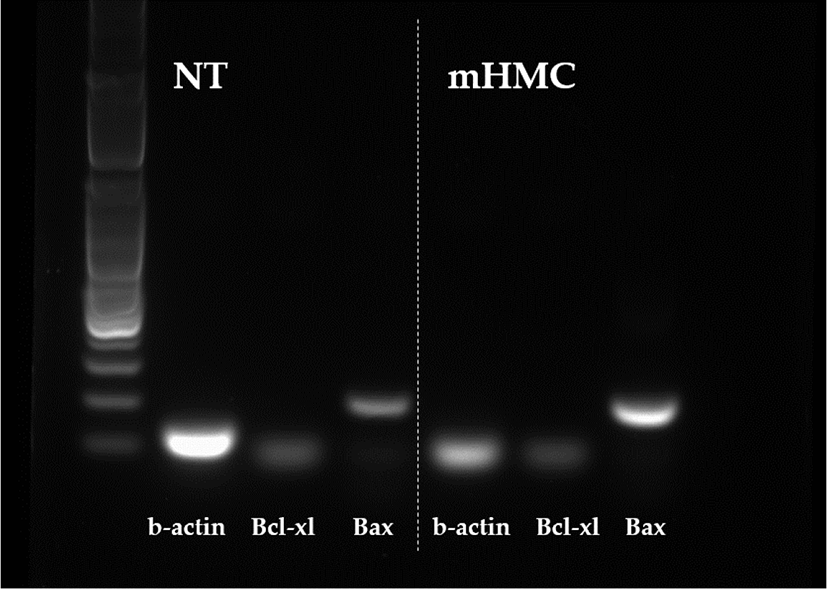
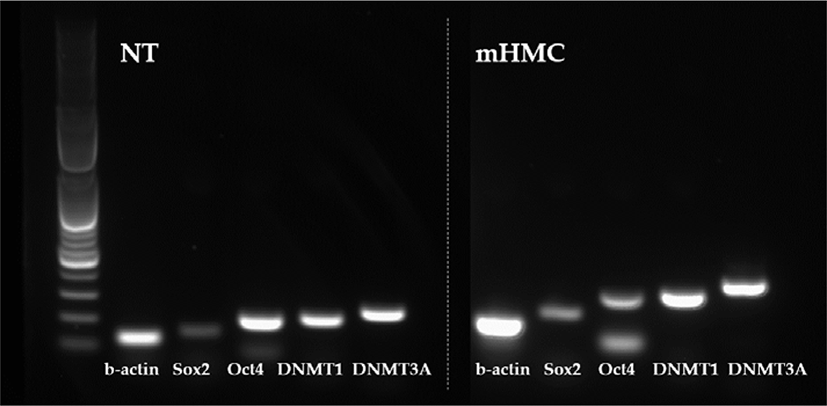
The expression of genes related to apoptosis (Bcl-xL; anti-apoptotic gene and Bax; pro-apoptotic gene), pluripotency (Oct4 [POU5F1], Sox2), and DNA methylation (DNMT1 and DNMT3α) were investigated. The primers used for investigation are specified in Table 1. Using the β-actin gene as a housekeeping gene, the abundance of each gene was investigated. Total mRNA was extracted from 30 blastocysts per group, using the RNeasy® Micro Kit (QIAGEN). Complementary DNA synthesis from extracted RNA was performed using the ExcelRT™ Reverse Transcription Kit (SMOBIO, Hsinchu, Taiwan). The reaction mixture (total 20 μL) consisted of 8 μL of total RNA, 1 μL of dNTP mix and 1 μL of oligo DT for denaturing, 4 μL of 5 × RT buffer, 4 μL of DEPC-treated H2O, 1 μL of RNase inhibitor, and 1 μL of Reverse Transcriptase as a first strand cDNA buffer. The mixture was subjected to PCR amplification (95°C for 4 min, followed by 40 cycles of 95°C for 30 s, 58°C for 30 s, and 72°C for 30 s, with a final extension at 73°C for 5 min). PCR products were electrophoresed in 1% agarose gels. A 100 base pair DNA ladder was used as a marker to check the molecular weight. Stained gels with loading dye were visible under UV light and the mRNA abundance was measured with the intensity of each band represented by the Image J program (Figs. 4 and 5).
All data were replicated at least three times, and statistical analyses were performed using two-way ANOVA, the Bonferroni post-test, and GraphPad Prism software version 5 (GraphPad, San Diego, CA, USA). Data were expressed as the mean ± SEM. A p-value < 0.05 was considered to indicate significantly different data.
RESULTS
As shown in Table 2, the rates of oocytes exhibiting cytoplasmic protrusions were significantly higher at 90 min than at 30 min. There was no significant difference among the groups treated with DEM for 60 min, 90 min, and 120 min. However, the cytoplasmic protrusions of the oocyte group treated with DEM for 60 min were difficult to identify without sucrose, and had to be checked under UV light to ascertain whether the oocyte nucleus was extruded with a part of the cytoplasm. The oocyte group treated with DEM for 120 min did not show a significant difference from the other groups. However, DEM treatment for 90 min was considered more efficient than DEM treatment for 120 min because of the reduced experimental time.
As shown in Table 3, the oocyte group treated with DEM after IVM showed a significantly higher proportion of cytoplasmic protrusions than the other groups. Similar to the previous finding that the oocytes treated with DEM for 90 min showed the highest rate of cytoplasmic protrusion, all oocyte groups in Table 3 were treated with 0.4 μg/mL DEM for 90 min.
As shown in Table 4, the accuracy of enucleation was significantly higher in the SCNT group than in the mHMC group (98.01 ± 0.57 vs. 83.83 ± 2.47, respectively). In addition, the number of surviving oocytes after enucleation was significantly higher in the SCNT group than in the mHMC group (96.50 ± 0.84 vs. 90.10 ± 2.11, respectively). The relative amount of removed cytoplasm after enucleation was approximately two times higher in the mHMC group than in the SCNT group.
The results of ROS levels are shown in Fig. 6. ROS levels increased after enucleation in both mHMC and SCNT groups, and no significant difference was observed.
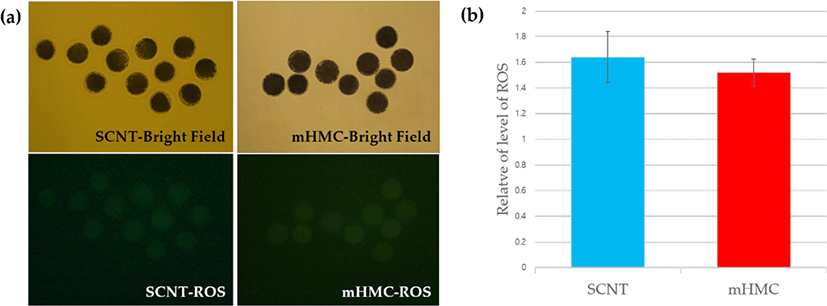
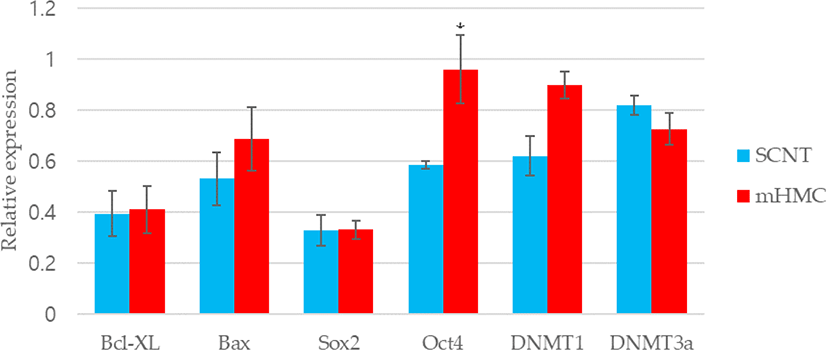
Table 5 shows that the in vitro developmental competence of embryos produced by SCNT and mHMC differed depending on the cloning method. The fusion rate was significantly higher in mHMC than SCNT (86.3 ± 2.44 vs 57.1 ± 5.87, respectively). The embryos at 8-cell and 16-cell stages showed significant differences (80.4 ± 2.31 vs 69.6 ± 5.37, in 8 cell, 54.9 ± 5.00 vs. 43.0 ± 3.75, in 16 cells, respectively). However, blastocyst rates did not show significant differences between mHMC and SCNT (17.9 ± 1.44 vs. 13.6 ± 1.99, respectively).
The expression of apoptosis and pluripotency-related genes (Bcl-xL; anti-apoptotic gene and Bax; pro-apoptotic gene) were evaluated by RT-PCR. The expression level of Bcl-xL was higher in mHMC embryos than in SCNT embryos, but with no significant difference. In addition, the expression of the pro-apoptotic gene Bax was higher in mHMC embryos than in SCNT embryos, but with no significant difference. The pluripotency gene Oct4 was significantly increased in mHMC embryos (p < 0.05), but Sox2 was similar between groups. DNMT1 and DNMT3α levels did not differ between the groups.
DISCUSSION
In this study, HMC was modified to simplify the cloning method by minimizing the loss of cytoplasm during enucleation using a pulled Pasteur pipet. The efficiency of enucleation and embryonic developmental competence were compared between mHMC and SCNT, for which a manipulator is essential. The embryos produced by SCNT and HMC exhibit similar replacement of the donor cell with their nucleus. However, the first difference is the existence of ZP. ZP has a role in protecting the oocyte from deleterious conditions in the culture medium and is an integral part of successful embryo development [24,25]. In this study, the existence of ZP did not have a significant impact on embryonic development. In a previous study, no significant difference in developmental competence was observed between zona-free and zona-intact embryos [26]. On the other hand, there were significant differences in enucleation efficiency. A cytoplasmic membrane is composed of phospholipid bilayers and can be recovered by fluid movement even with slight damage [27], however, it can be ruptured under high compressive stress [28]. Pulled Pasteur pipettes were made by hand by the researcher, therefore, they are wider in diameter than micromanipulator enucleation needles, which can further damage the cytoplasm by inconsistent pressure. In future studies, it will be necessary to standardize the pulled Pasteur pipette production process to reduce the pressure stress applied to the cytoplasmic membrane of oocytes.
In this study, DEM with a concentration of 0.4 μg/mL was applied for various times and oocyte statuses. A previous study reported that the ideal treatment time for DEM was 60 min [20], and added sucrose to increase osmoles in the medium in order to expand the perivitelline space in oocytes for easier observation of nucleus protrusions in mice and cows. However, porcine oocytes were not significantly affected by the presence or absence of sucrose [20]. In a previous goat study, oocytes treated with 0.8 ng/mL DEM for 30 min showed the highest protrusion rate. According to our results, the highest nuclear extrusion was observed in oocytes treated with DEM for 90 min without sucrose. The differences in the results of DEM concentration and treatment time may be due to the different species [29]. Our results showed that oocytes treated with DEM immediately after IVM showed a higher protrusion rate than other oocytes. DEM interferes with meiosis and is a microtubule depolymerizing agent, which destroys the 3D structure of the spindle, causing chromosome condensation [18], thus, for maternal chromosome protrusion, it is considered that the spindle must be stably arrested near the cytoplasmic membrane immediately after the first polar body is released. In addition, the oocytes were exposed to chemical stress during the removal of the cumulus cell and ZP, which could affect the stability of the oocyte. Therefore, we recommend treating DEM before denuding and after completion of the IVM stage to maintain the stable condition of oocyte cytoplasm.
ROS occur directly through the metabolic activity of the embryo [30] or environmental stress in in vitro conditions [31], causing adverse effects on the embryo. We assumed that the ROS level would be higher in mHMCs because the cytoplasm of oocytes without ZP is exposed directly in vitro in mHMC, unlike in SCNT. However, the ROS level in enucleated oocytes showed no significant difference between mHMC and SCNT, nor did the blastocyst formation rate. This study confirmed that the difference between mHMC and SCNT did not have a detrimental effect on enucleated oocytes.
Regarding the quality of blastocysts in mHMC and SCNT, we measured the expression levels of apoptosis-related genes, pluripotency-related genes, and reprogramming-related genes. The apoptotic pathway is maintained by the balance between the expression of pro- and anti-apoptotic genes in somatic cells [32,33]. The expression levels of Bcl-xl, an anti-apoptosis-related gene, and Bax, a pro-apoptotic gene, were measured in this study. Apoptosis is also observed in good and/or poor quality embryos during development [34]. In our results, high expression of Bax in mHMC was observed but was not significantly different between groups. The mHMC results confirmed that the oocytes were directly exposed to chemicals at various steps without the ZP, however, this did not have a detrimental effect on embryonic developmental competence.
We also investigated whether the expression levels of pluripotency-related genes Oct4 and Sox2 differed in mHMC and SCNT blastocysts. In many species, including pigs, this factor plays an important role in the preimplantation embryonic development stage [35]. The abnormal expression of Oct4 in porcine blastocysts derived from SCNT is related to low developmental competence of embryos. Thus, the abundance expression of the Oct4 gene indicates the developmental potential of embryos [36]. Sox2 expression is essential during embryogenesis and is the earliest marker of inner cells prior to ICM formation [37,38]. Although there was no significant difference in blastocyst formation rate between mHMC and SCNT, the expression level of Oct4 was significantly higher in the blastocysts of mHMC than in those of SCNT, and no difference was observed in the expression level of Sox2. These results confirmed that gene expression can vary depending on the cloning method.
DNA methylation is an important genetic marker that controls transcription. DNMT1 and DNMT3 α are enzymes that play a role in the transfer of methyl groups to cytosine nucleotides of the DNA sequence [39]. Although DNMT3 α encodes a DNA methyltransferase to perform de novo methylation, DNMT1 maintains the methylation pattern [40], which is one of the most important molecular activities in mammalian epigenetic gene regulation. Abnormal development of mammalian embryos is associated with aberrant activity of DNA methyltransferases. However, this study did not show any significant differences in methylation between the two groups. The results suggest that a difference in the cloning method did not have a significant influence on the methylation pattern of the embryos.
In conclusion, this study is the first to report porcine mHMC using the pulled Pasture pipette method instead of bisection to enucleate oocytes. The mHMC technique is preferable to the SCNT technique as it does not require the use of a micromanipulator and involves a simpler process. Moreover, the efficiency of enucleation and developmental competence of mHMC was found to be comparable to that of traditional SCNT. Thus, we demonstrate that porcine mHMC can be a cheaper and simpler alternative to SCNT.