
INTRODUCTION
Marbling is an important feature of beef quality [1]. It is particularly important in determining the price of beef in South Korea, Japan, and the United States because of its impact on tenderness, juiciness, and flavor [2]. Currently, with the rapid growth of high-grade beef demand in China’s domestic market, increasing intramuscular fat deposition has become a research hotspot in China’s beef industry [3]. Deposition of intramuscular fat is influenced by both genetic factors (such as breed, gender, and genotype) and non-genetic factors (such as castration, nutrition, stressors, and animal weight and age) [4,5]. Intramuscular adipocytes are formed by the lipid filling of fibroblastic cells within the perimysium, and marbling consists of fat cells distributed between muscle fibers [6–8]. In addition, the composition of fatty acids in meat, especially monounsaturated fatty acids (oleic and palmitoleic acids [POA]), is positively correlated with beef flavor [9].
Satellite muscle cells located between the myometrium and the basement membrane are pluripotent stem cell populations composed of Pax7-expressing progenitor cells [10]. They have mesenchymal plasticity, being able to commit either to myogenesis or to a mesenchymal alternative differentiation program [11]. Earlier studies have shown that cells from myogenic cultures that have showed myogenic response could be transferred from this pathway when treated with bone-morphogenetic proteins or adipogenic-inducing agents [12,13]. Our previous studies have shown that oleic acid, as the most abundant monounsaturated fatty acid in beef marbling, has the ability to differentiate microsatellite cells into adipocyte-like lipid droplets [14].
However, it is not clear whether POA, the second most abundant monounsaturated fatty acid in beef marbling, has the same characteristics as oleic acid. Therefore, we hypothesized that treatment of bovine skeletal muscle satellite cells with POA can induce the differentiation of muscle satellite cells into adipocytes, thereby promoting the formation of intramuscular fat and improving the quality of beef.
MATERIALS AND METHODS
All experimental procedures involving animals were conducted using the approval code YBU-20160303 provided by the Institutional Animal Care and Use Committee of Yanbian University.
Bovine satellite cells (BSCs) were isolated from the semimembranosus muscle of six 18-month old Yanbian Yellow cattle raised at Yanbian University. The BSCs were excides from the muscle tissue samples as previously described [14]. In brief, the muscle samples were minced and digested using 1 mg Pronase (10165921001, Roche, Basel, Swiss) in 1 mL of Earl’s Balanced Salt Wash (EBSS, Sigma-Aldrich, St. Louis, MO, USA). After digestion, the mixture were subjected to differential centrifugation to collect the mononucleated cells, which were suspended in Dulbecco’s Modified Eagle’s medium ([DMEM] Gibco, Thermo Fisher Scientific, Waltham, MA, USA) that contained 10% fetal bovine serum ([FBS] Gibco, Thermo Fisher Scientific), 10% (v/v) dimethyl sulfoxide ([DMSO] Sigma-Aldrich) and 1× antibiotic-antimycotic (Gibco, Thermo Fisher Scientific). The cells were frozen and stored in liquid nitrogen for subsequent studies.
The BSCs were cultured in growth media composed of DMEM, 10% FBS, 1% penicillin/streptomycin at optimal cell growth conditions in incubator (38.5°C, 95% O2, and 5% CO2). The cell cultures were fed with growth medium every 48 h. Cell cultures that reached 80%–90% confluence were induced to differentiate by switching from growth medium to a differentiation medium comprised of DMEM, 5% horse serum (HS), and 1% penicillin/streptomycin. The differentiating or proliferating BSCs were exposed to different concentrations of POA (50 μM; 100 μM; and 200 μM, Sigma-Aldrich) or to none-POA (control cells) in media for 96 h. POA was dissolved in ethanol (100 mmol/L) prior to its addition to the cell cultures [15]. To measure the viability and size of the cells, the BSCs were harvested and stained with trypan blue staining for measurement Luna automatic cell counter (Logo Biosystems, Anyang, Korea). The experiment was conducted in triplicates or in three independent incubations.
The cell proliferation was determined by MTT assay using the Cell Proliferation Reagent Kit (MTT ST316, Beyotime, Shanghai, China). The BSCs were seeded in 96-well plates at a seeding density of 2 × 103 cells and maintained for 24 and 48 h in 10%-FBS growth media. The cells were then incubated with 5 mg/mL MTT in phosphate buffered saline (PBS) for 4 h, after which the formazan crystals were dissolved by adding 150 μL DMSO to each well where the plates were shaken for 10 minutes to fully dissolve the formazan crystals. The absorbance was measured using a microplate reader at 490 nm wavelength (reference wavelengths 450 nm, 570 nm). The results were obtained and used to draw the cell growth curve with as abscissa and absorbance as the ordinate.
To verify whether the cells isolated were BSC, the primary cells were stained for immu-nofluorescence. Cultured satellite cells were fixed with ice-cold 4% paraformaldehyde for 20 min, rinsed twice with PBS, and permeated with 0.2% Triton x-100 (in PBS) at room temperature for 30 min. The permeabilized cells were blocked with PBS containing 5% bovine serum albumin (BSA) and shaken at room temperature for 1 h, and incubated with primary antibodies: polyclonal anti-myogenic differentiation 1 (MYOD1; BS-2442R, Bioss, Woburn, MA, USA), anti-Pax7 (AB-528428, Abcam, Cambridge, UK), and anti-desmin Po (BS-20702R, Bioss) in 5% BSA at 1:100 dilutions for 2 h at room temperature. After washing with PBS, the cells were incubated with HRP-labeled Goat Anti-Mouse IgG (H + L) (A0216, Beyotime, Shanghai, China) or Goat Anti-rabbit IgG-HRP antibody (BS-3550R, Bioss) for 2 h at room temperature in dark room and mounted in 0.1 μg/mL DAPI solution for 30 min. The staining of the cells was observed using a fluorescence microscope (WF10X, Olympus, Tokyo, Japan) afterwards.
The Oil-Red-O was used to confirm and quantify the accumulation of lipid droplets in differentiated BSCs treated without or with POA. The specific staining process was performed according to the instructions provided by Oil Red O Staining Kit (G1262, Solarbio, Beijing, China). Briefly, the cells were fixed with 10% neutral buffer formalin, after which the cultures were stained with 0.5% Oil-Red-O solution in darkness. The adipocytes were identified by the presence of lipid droplets. Images of the lipid droplets were taken using collected using an inverted bio microscope (IX-73, Olympus). Images of intracellular lipid droplets stained using the Oil Red O kit were analyzed by computer image using the ImageJ software (https://imagej.nih.gov/ij/) and the method of analysis described by Deutsch et al. [16] was followed. Briefly, the 8-bit red–green–blue image was threshold converted into a binary image consisting only the pixels that represent the lipid droplets, after which the images were watershed object separated to identify the borders of adjacing lipid droplets, and the scale of the images was set. The amount and individual size of the lipid droplets in the image presented by ImageJ as surface area in square micrometers (µm2) were measured thereafter.
The triacylglycerol (TAG) in culture cells was measured using the TAG assay kit (Applygen Technologies, Beijing, China). The cells were washed with PBS three times to remove residual medium, and were then lysed with RIPA lysis buffer (10 min). Accumulated triglyceride was digested to release the TAG into the lysate and aliquots were removed and the amount of glycerol liberated from TAG was determined by spectrophotometric detection at 570 nm and is linear over a glycerol concentration of 0–500 μM.
The adiponectin in BSC culture media was analyzed using a bovine adiponectin ELISA kit (Mlbio, Shanghai, China), and the analysis was conducted by following the manufacturer’s protocol. The adiponectin concentration was measured with a microplate reader (iMark, BIO-RAD, Hercules, CA, USA) at 450 nm, and a reference curve was generated using reference standards of known concentrations of adiponectin in the same assay. The reference curve was used to calculate the amount of adiponectin in the samples. The mean interassay precision was < 8.4%, and the intraassay precision was < 5.7% for the adiponectin assay.
Total RNA was extracted from cells incubated for 96 h using Trizol reagent (Thermo Fisher Scientific) according to the manufacturer’s instruction. 1 μg of RNA from each treated cell was reversed transcribed to cDNA using a FastKing one-step method (Tiangen Biotech, Beijing, China) according to the manufacturer’s protocol. The gene expression levels were determined using the appropriate forward and reverse primers and performed using the SuperReal PreMix Plus (SYBRGreen, Tiangen Biotech) with the Agilent Fast Real-Time PCR System using). The sequences of the primers are given in Table 1, and the target genes were as follows: paired box 3 (Pax3) and paired box 7 (Pax7), myogenic differentiation antigen (MYOD), myogenin (MYOG), myofactor-5, and myofactor-4 (MYF5, MRF4) are the myogenic-specific marker genes selected to determine the degree of myogenesis in cultured BSCs; peroxisome proliferator-activated receptor gamma (PPARγ), CCAAT/enhancer-binding protein alpha (C/EBPα) and beta (C/EBPβ), G-protein coupled receptor 43 (GPR43), sterol-regulated binding protein 1 (SREBP1), and stearoyl coenzyme-A desaturation enzyme (SCD), carnitine palmitoyltransferase 1 (CPT1), AMP-activated protein kinase alpha-1 (AMPKα), fatty acid synthase (FASN), lipoprotein lipase (LPL), glycerin-3-phosphate acyltransferase 3 (GPAT3), lipid droplet formation-related protein, perilipin (PLIN1), adipose differentiation-related protein (adipophilin; PLIN2), and tail-interacting protein of 47 kDa (TIP47/PLIN3) were the adipogenic/lipogenic gene markers used to analyze the differentiation of BSCs into adipocytes at different concentrations of the POA. The thermal cycling parameters were as follows: pre-denaturation of samples at 95°C for 15 min, then amplification for 40 cycles, which consisted of 10 s at 95°C, 30 s at 55°C and 30 s at 72°C, with a melting curve from 65°C to 95°C. The 2−△△ct method was used to determine the relative fold-changes and all data were normalized with the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
PCR, polymerase chain reaction; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; Pax3, paired box 3; Pax7, paired box 7; MYOD, myogenic differentiation antigen; MYOG, myogenin; MYF5, myofactor-5; MRF4, myofactor-4; PPARγ, peroxisome proliferator-activated receptor gamma; C/EBPα, CCAAT/enhancer-binding protein alpha; C/EBPβ, CCAAT/enhancer-binding protein beta; SREBP1, sterol-regulated binding protein 1; GPR43, G-protein coupled receptor 43; SCD, stearoyl coenzyme-A desaturation enzyme; CPT1β, carnitine palmitoyltransferase 1 beta; ACSL3, acyl-CoA synthetase long-chain 3; AMPKα, AMP-activated protein kinase alpha-1; LPL, lipoprotein lipase; FASN, fatty acid synthase; GPAT3, glycerin-3-phosphate acyltransferase 3; PLIN1, lipid droplet formation-related protein, perilipin; PLIN2, adipose differentiation-related protein (adipophilin); PLIN3, tail-interacting protein of 47 kDa (TIP47); ADI, adiponectin.
Equal amounts of protein samples (30 µg/sample) were separated electrophoretically by 10% sodium dodecyl sulphate (SDS)-polyacrylamide gel electrophoresis (PAGE), blotted onto a PVDF membrane (Millipore, Billerica, MA, USA), and probed with primary and secondary antibodies. Primary antibodies used were Desmin (bs-20702R), PPARγ (bs-4509R), C/EBPα (bs-1630R), MYOD (bs-2442R), and MYOG (bs-3550R), and the secondary antibodies were horseradish peroxidase-labelled goat anti-mouse or anti-rabbit IgG (bs-0295G) obtained from Bioss, while anti-Pax7 (AB-528428) and GAPDH (ab9484) were procured from Abcam.
The data in this study were all presented as mean ± SEM. Significant differences among the treatments were analyzed using one-way analysis of variance and Post Hoc Tukey’s multiple comparison test GraphPad Prism 6.07 (GraphPad Software, La Jolla, CA, USA) at significance level of p < 0.05.
RESULTS
The average cell size in CON + 100 µM POA ([POAM] 20.60 ± 0.50 µm) and CON + 200 µM POA ([POAH] 23.36 ± 0.39 µm) treatments was higher than that of CON (15.93 ± 0.27 µm) (p < 0.001), whereas there was no significant difference between CON (15.93 ± 0.27 µm) and CON + 50 µM POA ([POAL] 17.20 ± 0.20 µm) (Fig. 1A). However, there was no difference in cell viability among the treatments (p = 0.9053; Fig. 1B).
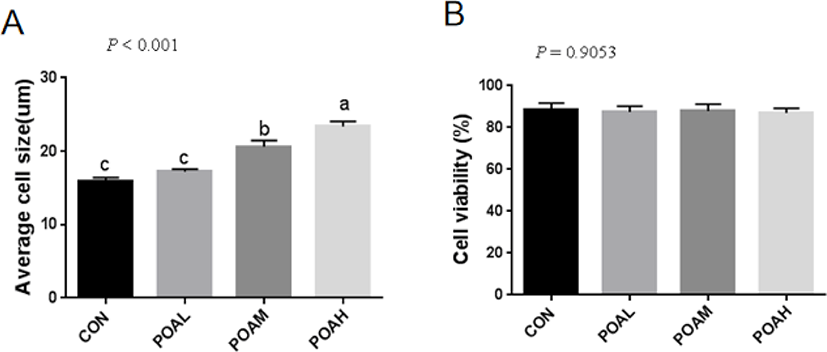
Fig. 2A shows the formation of myotubes and the multinucleate nature of cells at 6 days post-confluence. We examined the expression of the myogenic markers MYOD and desmin. Pax7, in particular, was considered to be a marker of muscle satellite cells. Pax7, MYOD, and desmin were detected in the nuclei of both mononucleate and fused cells (Fig. 2B).
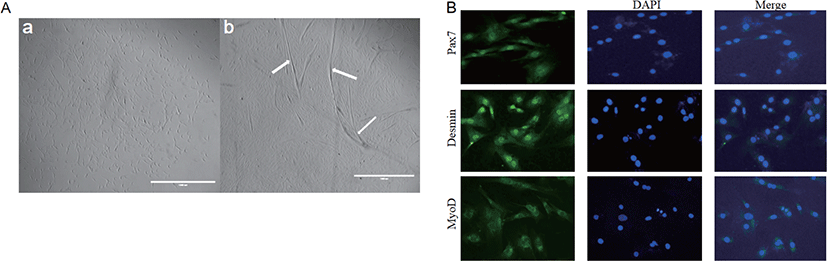
The adipogenic differentiation of BSC was induced by adding POA to the differentiation medium with 5% HS. Oil red O staining was positive after 96 h of differentiation culture. Cells added to the POA group gradually developed into adipocyte-like cells, which did not occur in cells in CON. Moreover, with the increase of POA concentration, the area of lipid droplets increased significantly (Fig. 3A). The adiponectin concentration in the medium also increased with the POA concentration (Fig. 3D).
Compared with CON, different concentrations of POA significantly increased intracellular TAG content and lipid droplet area (Fig. 3). And the higher the concentration of POA added, the higher the intracellular TAG content (Fig. 3C), and the lipid droplet area also increased significantly (Fig. 3B).
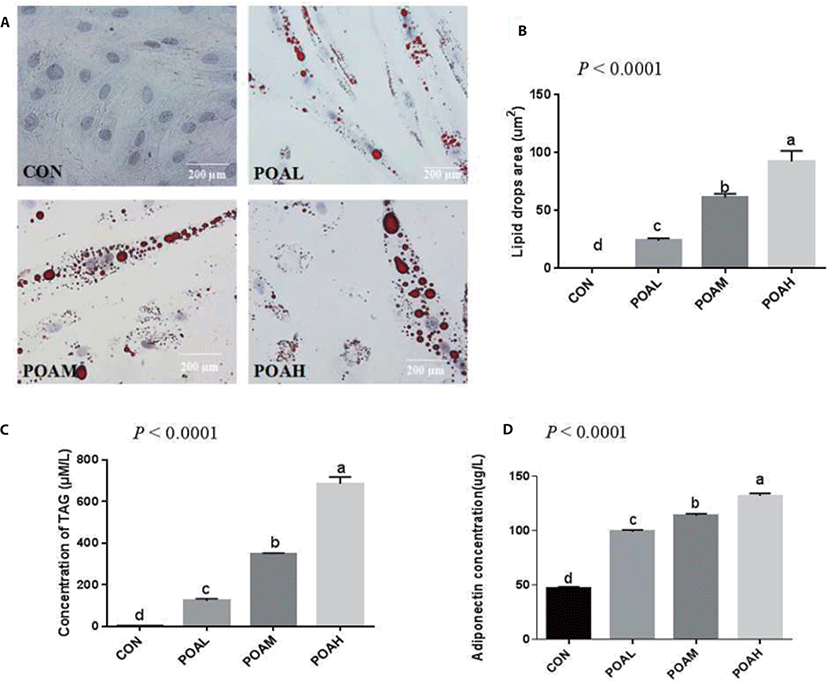
Compared with CON, POA showed increased expression of Pax3 (p < 0.0001; Fig. 4A) and Pax7 (p < 0.0001; Fig. 4B) genes. Different concentrations of POA significantly inhibited the expression of myogenic genes MYOD, MYOG, MYF5, and MRF4 (p < 0.0001; Fig. 4C, D, E, and F). Western blot analysis was used to determine the expression levels of PAX7, MYOD, and MYOG. Results showed that POA treatment increased the protein expression of myogenic transcription factor PAX7 and decreased the MYOD and MYOG in BSC (Fig. 4G).
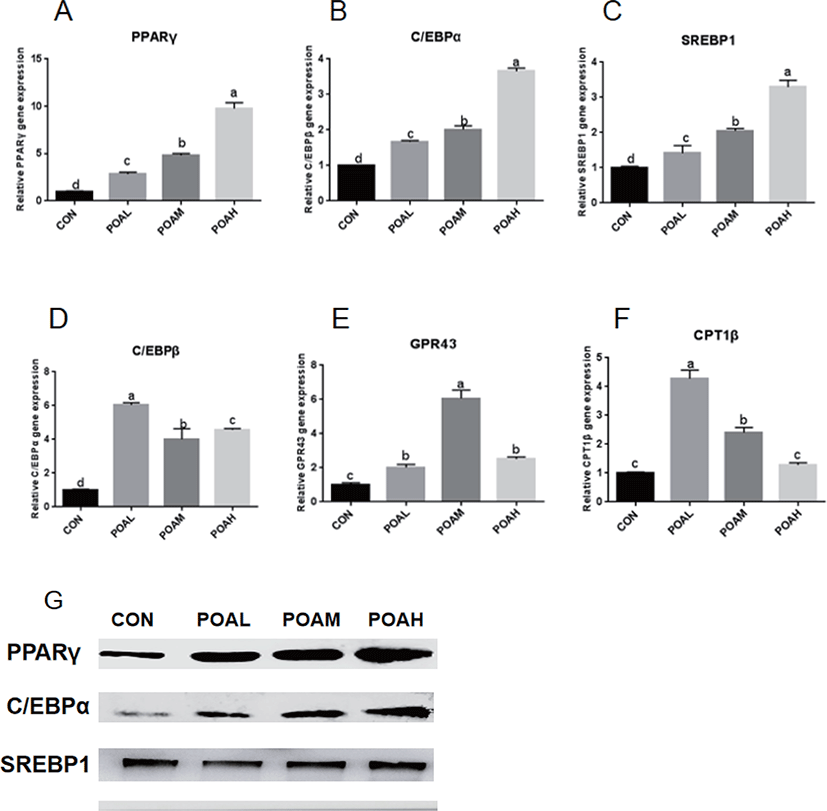
POAL, POAM, and POAH treatments showed increased PPARγ, C/EBPα, SREBP1 (p < 0.0001; Fig. 5A, B, and C) expressions compared with CON, and POAH treatment showed the highest increase. POA treatment also promoted the expression of C/EBPβ, GPR43, and CPT1β (p < 0.0001; Fig. 5D, E, and F) and other adipogenic genes. Western blot analysis was used to determine the expression levels of PPARγ, C/EBPα, and SREBP1 and POA treatment increased the protein expression of adipogenic transcription factors PPARγ, C/EBPα, and SREBP1 in BSC as shown in Fig. 5G.
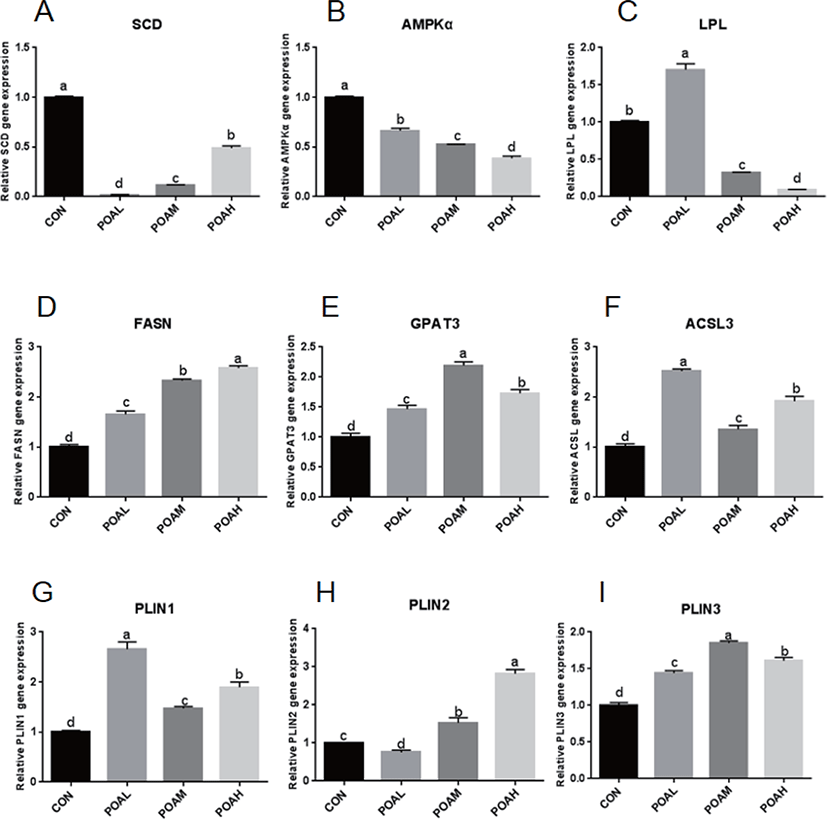
Expression of SCD and AMPKα was lower in the POA treatment than in CON (p < 0.0001; Fig. 6A and B); the expression of LPL was lower in the POAM and POAH treatments than in CON (p < 0.0001; Fig. 6C). However, the expression of genes related to fatty acid synthesis such as FASN, GPAT3, acyl-CoA synthetase long-chain 3 (ACSL3) was higher in THE POAM and POAH treatments than in CON (p < 0.0001; Fig. 6D, E, and F). Moreover, the expression of PAT family genes was positively correlated with the increasing concentrations of POA (p < 0.0001; Fig. 6G, H, and I); and POA also promoted the expression of CD36, Leptin, and Adiponectin, which are closely related to lipid synthesis (Fig. 6J, K, L, and M).
DISCUSSION
The purpose of this study was to investigate the effect of POA on the differentiation of bovine BSCs. Previous studies have shown that BSCs can be isolated from adult Hanwoo muscle, and that BSCs can proliferate and be induced to differentiate into adipocytes [17]. Xu et al. [18] reported that all fatty acids did not affect the proliferation of bovine muscle-derived satellite cells (MDSC) through the study of proliferation rates and cell cycle progression, so we hypothesized that POA could promote the differentiation of BSCs. During the growth of skeletal muscle, the number of BSCs accounts for approximately 80% of the myonuclei. These numbers decrease to 4%–15% of the total nuclei in the muscle fiber as the animal approaches its mature size [19]. This decrease in BSCs is generally accompanied by cell differentiation or the formation of new muscle fibers [20]. In our experiment, after treating BSCs with different concentrations of the POA, there was no difference in the viability of BSCs, but a significant difference in cell size. We speculate that this may be caused by changes in cells during differentiation. Neutral lipids in skeletal muscle are mainly stored in lipid droplets in the form of TAGs [21]. We performed Oil Red O staining and determined intracellular TAG concentration after the 96-h incubation of BSCs treated with the POA. The results of Oil Red O positive staining and the significant increase of TAG concentration confirmed our hypothesis that POA promoted the muscle satellite cells differentiation into adipocytes.
It has been shown that oleic acid, an 18-C unsaturated fatty acid, promotes adipose differentiation by up-regulating adipocytes transcription factor PPARγ, C/EBPα, and C/EBPβ [14,22,23]. According to reports, BSCs can be transferred to adipocytes by using PPARγ agonists [13,24,25]. In this study, PPARγ, C/EBPα, C/EBPβ, and other adipocytes transcription factors expressions significantly increased in BSCs differentiated by the POA. In addition, the lipid metabolism factors CPT1β and GPR43 were up-regulated, but the expression of SCD and AMPKα were significantly inhibited, which was consistent with the research of [26,27]; that is, the inhibition of SCD catalytic activity reduced the activity of genes associated with de novo fatty acid synthesis, while increasing the expression of genes related to fatty acid oxidation, such as CPT1β. AMPK is an important cellular energy sensor, which regulates metabolism and energy balance [28]. A large number of studies have shown that AMPK regulates the expression of polyester-related genes (e.g., PPARγ, C/EBPα, FAS, SREBP-1) in fat precursor cells differentiation through Wnt/β-catenin, mTORC1, MAPK, and SIK signaling pathways [29,30]. In skeletal muscle, AMPK activation can increase sugar intake, enhance fat oxidation, and reduce TAG synthesis through phosphorylation of ACC2 [31]. Although the increased expression of CPT1β and other fatty acid oxidation-related genes induced by BSCs treated with POA may inhibit the aggregation of TAG and the formation of lipid droplets, the increase of TAGs in muscle is mainly caused by the imbalance between fat deposition and utilization ([32–35]).
In this study, although the POA treatment group up-regulated the expression of Pax3 and Pax7, it significantly down-regulated the expression of myogenic regulatory factors MYOD, MyoG, Myf5, and MRF4. Chung et al. [36] demonstrated that treating muscle cells in culture with 100 µM oleic acid and other growth factors down-regulated myogenin gene expression. The decreased expression of these myogenin genes suggests that the development of muscle fibers is inhibited.
Among TAGs-containing lipid droplets, a sub-population expands after their initial formation. TAG is locally generated by a series of enzymes such as GPAT3 and diacylglycerol O-acyltransferase 2 (DGAT2), which leads to linear volume expansion over time of these lipid droplets specifically [37]. The PAT-family proteins are a class of small protein molecules in lipid metabolism. Positioned at the lipid droplet surface, PAT proteins manage access of other proteins to the lipid esters within the lipid droplet core and can interact with cellular machinery important for lipid droplet biogenesis. PAT-family proteins are not directly involved in the synthesis and decomposition of lipids [38].
PLIN2 is present in the early stage of lipid droplet formation, promoting the formation of lipid droplets, and it is replaced by PLIN1 when the lipid droplets mature. Interestingly, the expressions of PLIN2 and PLIN1 are activated or inhibited by the co-activator PPARγ [39,40]. Adipose differentiation-related protein (ADRP) and perilipin are markers of cellular lipid accumulation [41,42]. Imamura et al. [43] showed that ADRP stimulated lipid accumulation and lipid droplet formation in murine fibroblasts by transporting endogenous free fatty acids from the cytosol to the surface of lipid droplets without induction of other adipocyte-specific genes or other lipogenic genes in murine fibroblast cells. Our study confirmed that after 96-h incubation of BSCs treated with POA, the gene expression of PAT-family proteins (PLIN1, PLIN2, and PLIN3) was significantly up-regulated, which was closely related to the formation of lipid droplets after differentiation, and further confirmed that BSCs can transdifferentiate into adipocytes.
In this study, POA significantly up-regulated the expression of leptin and adiponectin genes, especially in the POAH, and the concentration of adiponectin in the medium increased significantly. Leptin and adiponectin are two of the most important markers of adipocyte differentiation. Leptin is considered a pleiotropic hormone whose metabolic functions include appetite control, energy balance, and regulation of liposugar metabolism [44,45]. Adiponectin may be produced exclusively by adipocytes and plays a role in regulating insulin sensitivity [46–48]. As a fatty acid transporter, the main function of CD36 is to transport long-chain fatty acids, which can promote the absorption of free fatty acids in adipocytes, increase intracellular lipid accumulation, expand adipocyte volume, and also promote the transformation of preadipocytes into mature adipocytes, increasing the number of adipocytes [49,50]. This is consistent with the significant difference between the number of lipid droplets and the area of lipid droplets in cells with increasing POA concentration observed in our study.
In conclusion, treatment of BSC with POA can promote the transdifferentiation of BSCs into adipocytes where at least a portion of BSCs were converted into adipocytes as evidenced by the downregulations of myogenic related genes that caused the depression of muscle fiber development, and upregulation of adipogenic related genes that caused the transdifferentiation of muscle cells into adipocyte-like cells. This study demonstrated that POA was could be a positive regulator for differentiation of BSCs to ADPs. Our results suggest that POA could be useful in increasing the marbling content of beef to improve meat quality and this also shows that bovine satellite cells could be useful study models in the development of meat culture technology.