













The well-being and health status of economic animals are more deteriorative due to the intensive farming practices. Feed-additive probiotics including lactic acid bacteria and Bacillus spp. are commonly used to modulate host health and improve performance in pig production [1,2]. Typically, a culture-dependent isolation coupled with in-vitro characterization methods have been used to establish probiotic potential of the strains. Due to rapid development of next generation sequencing in the past decade, scientists are now exploring whole genome sequencing to identify and functionally characterize novel probiotic strains. A number of potential probiotic strains have been identified based on their putative functional genes from their whole genome [3].
Paenibacillus spp. are well known as growth promoters for crops, but there are very limited studies exploring their probiotic potentials, despite the fact that they can produce diverse kinds of active substances namely antimicrobial peptides (bacteriocin and lipopeptide), volatile organic compounds, and digestive enzymes (amylase, cellulase, lipase, protease, etc), among many others [4].
Previously, we have isolated a novel Paenibacillus strain SK3146T (= KACC 18876T = LMG 29568T) from a pig feed, which was taxonomically assigned as Paenibacillus konkukensis sp. nov. [5]. In this study, we provide a detailed description of the complete genome sequence of SK3146 and analyzed its putative functional genes related to digestive enzymes and bacteriocin which could be beneficial attributes as a functional feed additive.
SK3146 was cultured in Luria-Bertani broth for two days at 37°C under shaking conditions (100 rpm). Genomic DNA of SK3146 was extracted using the Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA) according to the manufacturer’s instructions. The genome of SK3146 was completely sequenced using the PacBio® RS II system by Macrogen (Seoul, Korea). The PacBio RS II system libraries were prepared using the SMRTbell template prep kit v 1.0. In total, 142,242 bp PacBio subreads with 1,041,901,553 bp were generated using the PacBio® RS II system, and their mean length and N50 value were 7,324 and 10,697 bp, respectively. The sequencing reads were de novo assembled using the HGAP analysis with default options. The assembly was completed with the PacBio RS II system. Annotation of coding DNA sequence (CDS) and functional genes were analyzed by the Prokka v1.10. The general features of the SK3146 were analyzed based on its complete genome sequence using the Geneious 8.1.9 software (Biomatters, Auckland, New Zealand) [6]. The predicted CDS were classified depending on the clusters of orthologous genes (COG), followed by the construction of a circular genome map and analysis of protein function by a web server: Bacterial Annotation System (https://www.basys.ca/) [7].
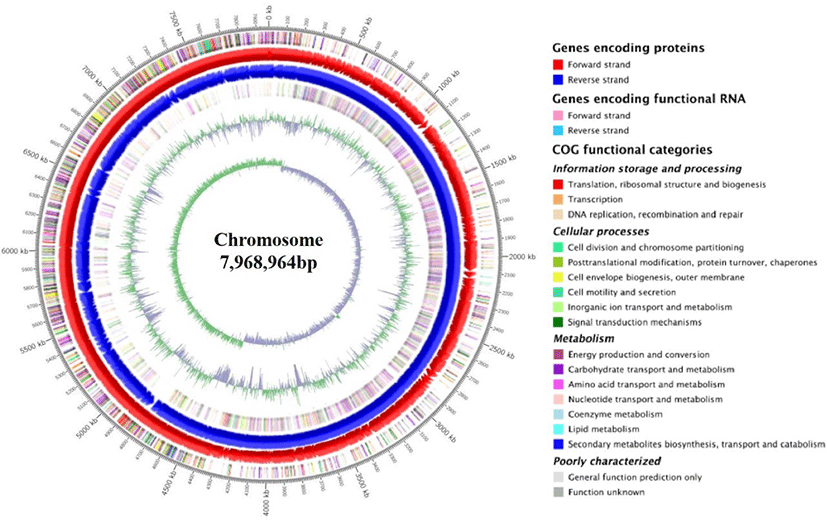
The circular genome visualization and general features of SK3146 genome are presented in Fig. 1 and summarized in Table 1, respectively. The complete genome of the strain consists of a single circular chromosome measuring 7,968,964 bp in size and 53.4% in guanine + cytosine (G+C) content. A total of 6,988 genes were predicted in the genome including 6,842 CDS, 37 ribosomal RNAs (rRNA), 108 transfer RNAs (tRNA), and 1 transfer-messenger RNA (tmRNA) loci. In addition, 10 clustered regularly interspaced short palindromic repeats (CRISPR) elements, three prophage regions, and 16 insertion sequence (IS) elements were identified in the genome.
Furthermore, we have analyzed the presence of potential enzymes in the genome of SK3146 via protein function annotation with the Kyoto Encyclopedia of Genes and Genomes database, which provides specific substrates, reactions, and enzyme nomenclature [8]. We then categorized the genes encoding potential enzymes of SK3146 according to enzyme code number. The class of hydrolases (EC3) including galactosidase, glucosidase, cellulase, lipase, xylanase, protease, and others in SK3146 are listed in Table 2. EC3 hydrolases including phosphatases, glycosidases, peptidases, nucleosidases, and lipases are widely used in feed additive industry to improve digestibility and bioavailability of nutrients in animal feeds [9]. Besides enzyme encoding genes, one bacteriocin encoding gene was also found on the chromosome of SK3146. The hydrolytic enzymes such as glucanase, cellulase, protease, and chitinase of Paenibacillus have been reported to have anti-fungal activities via destruction of fungal cell wall [10]. Moreover, β-glucosidase, cellulase, xylanase, and protease have been demonstrated to reduce carbohydrate- and protein-based anti-nutritional factors present in the plant-derived protein sources and consequently improving the nutritional quality of feed [11].
In the present study, the complete genome of P. konkukensis sp. nov., SK3146 isolated from a pig feed has been reported. The genome of SK3146 encodes multiple enzymes that could be applied to improve the digestibility and bioavailability of nutrients of animal feed. A gene encoding bacteriocin was also identified. Thus, the genome mining conducted in this study suggests that stain SK3146 has significant potential as a probiotic for use in feed additive applications. In addition, the genome information of SK3146 widens our understanding on the whole genus of Paenibacillus to explore and develop next generation probiotics. The genome-based protein prediction will be validated by in vitro characterization and in vivo animal study in near future.