
INTRODUCTION
The Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR-associated protein 9 (CRISPR/Cas9) system is a revolutionary gene-editing technology that involves Cas9 and a single guide RNA (sgRNA) [1]. Cas9, an endonuclease, acts as molecular scissors and cuts the target DNA. The sgRNA is a short RNA molecule that guides Cas9 to a specific target DNA sequence and induces deletions or insertions (indels) of base pairs at the target site. This gene-editing strategy is more advantageous than previous ones, including homologous recombination and the Zinc Finger Nuclease technique, as it can simultaneously alter multiple target genes or genomic loci. Hence, CRISPR/Cas9 has been widely used in animals, including mice [2, 3], rats [4], chickens, and pigs [5-10].
Pigs are recognized for their agricultural significance as a meat source for humans and in the laboratory as an alternative animal model for biomedical research. Pigs are highly valued because they produce numerous offspring and have a fast life cycle, making them efficient in breeding and meat production. Moreover, pigs share similar characteristics with humans in various aspects, including their immune system, metabolic functions, and other physiological as well as anatomical features [11]. Several gene-edited (GE) pigs have been generated for various applications, including farm production [9, 12], xenotransplantation [10,13,14], and modeling of human diseases [14,15].
However, generation of GE pigs is an inefficient, extremely laborious, and time-consuming process compared to that of mouse models [16–19]. It involves: 1) selecting a target candidate gene to be modified, 2) generating transgenic embryos by somatic cell nuclear transfer (SCNT) or by direct injection of Cas9 protein and sgRNA, and 3) transferring transgenic embryos into surrogates, followed by caring for the pregnant pigs and their offspring. In particular, if the developed pigs exhibit characteristics that differ from those expected, they cannot be used for the original research purpose, and the entire process must be performed again from the beginning. Therefore, it is crucial to select promising target genes that function as expected in vivo before generating GE pigs.
To address this issue, in vivo gene editing has been developed as an approach that allows assessment of gene function in living animals. Several studies on in vivo gene editing by delivering Cas9 protein and sgRNA into live animals have been reported [20–28]. However, the overall editing efficiency remains low because of the limited packaging capacity of the lentivirus and adeno-associated virus (AAV) for delivering the Cas9 endonuclease gene. [29–31]. To overcome this, mouse models expressing Cas9 have been developed, allowing gene editing to be achieved by simply injecting sgRNA [32–35]. In pigs, multiple Cas9-expressing lines have been reported [36–39]. In this study, we created a Cas9-expressing pig via precise knock-in of the Cas9 gene cassette into the ROSA26 locus and validated in vitro gene editing using primary cells derived from the pig. This pig line can be used to study gene function more precisely, facilitating the generation of utilizable GE pigs for both agricultural and biomedical fields.
MATERIALS AND METHODS
The experimental protocols were reviewed and approved by the Institutional Animal Care and Use Committee of the National Institute of Animal Science, RDA (IACUC No. NIAS2021-0509). Landrace pigs were used as surrogate mothers and were purchased from local farms.
Porcine ear fibroblasts (pEFs) isolated from Nanchukmacdon, a pig breed derived from Korean native black pigs , were used for the gene-targeting experiments [40]. The pEFs were cultured in DMEM (high glucose, GlutaMAX™ Supplement, pyruvate, Gibco, Grand Island, NY, USA) supplemented with 15% fetal bovine serum (Gibco), 1× MEM Non-Essential Amino Acids (Gibco), 1× antibiotic-antimycotic (Gibco), 1× LSGS, and 1× β-mercaptoethanol (Gibco,), at 37°C in an incubator maintaining 5% CO2.
For precise integration of the Cas9 gene at the porcine ROSA26 locus, we generated a Cas9 gene cassette. For this purpose, two separate vectors were synthesized; one vector consisted of a 0.8-kb 5′ homology arm (HA) corresponding to a region of porcine ROSA26 intron 1, organized as follows: a 0.1-bp fragment harboring viral splice acceptor (SA), LoxP 2272 and LoxP sequences, an FRT sequence, a PGK promoter-driven neomycin resistance gene and poly-A signal, FRT sequence, and a 0.8-kb 3′ HA corresponding to a region of porcine ROSA26 intron 1. The other vector comprised a ~5.6 kb Kozak sequence, Cas9 gene, nucleoplasmin NLS, P2A, and tdTomato gene cassette. The two vectors were digested using AvrII and KpnI, and the essential fragments from each vector were ligated together. We obtained the final pRosa26-Cas9 gene donor vector containing a 0.8-kb 5′ HA and 3′ HA, a viral SA, SpCas9-P2A-tdTomato sequence flanked by loxP and mutant loxP2272 sites in different orientations. These sites enable inversion of the SpCas9-P2A-tdTomato expression cassette following Cre-mediated recombination and cease SpCas9-T2A-tdTomato expression. Additionally, the vector included an SV40 polyadenylation (poly-A) signal sequence and a PGK promoter-driven neomycin resistance gene flanked by FRT sites. These FRT sites facilitated removal of the neomycin gene expression cassette through flippase-mediated recombination.
The cells, cultured in a collagen-coated 35-mm dish, were co-transfected with 0.5 μg of linearized Cas9 donor vector, 1.2 μg of ROSA26-targeting sgRNA, and 6.25 μg of Cas9 protein using Lipofectamine 3000 and CRISPRMAX transfection reagents (Thermo Fisher Scientific, Waltham, MA, USA) following the instructions provided by the manufacturer. After 2 days of transfection, the transfected cells were divided into five 100-mm culture dishes using a series of five consecutive 10-fold serial dilutions and selected through incubation in 500 μg/mL geneticin (Gibco) for 1 day and subsequently using 1000 μg/mL geneticin. After 7–21 days of selection, geneticin-resistant colonies were isolated using cloning cylinders and were cultured in collagen-coated 24-well plates using the modified DMEM media described in the previous section. After achieving 80%–100% confluency, the cells were transferred and cultured in 6-well plates until they reached 100% confluency. One-third of the cells were used for identifying the targeted clones through 5’- and 3’-junction polymerase chain reaction (PCR) genotyping analysis, while the remaining two-thirds of the cells were cryopreserved in liquid nitrogen. After the identification process, the positive cryopreserved cells were expanded and cryopreserved for SCNT.
To produce cloned porcine embryos, Cas9-expressing cells were subjected to SCNT, which was performed following the previously described protocols with slight modifications [41–42]. Briefly, ovaries from prepubertal gilts were procured from a local slaughterhouse to obtain immature oocytes. Follicular fluid containing cumulus-oocyte complexes (COCs) was aspirated from follicles measuring 3–6 mm in diameter and subsequently washed three times with TCM199 media with HEPES (#12340-030, Gibco) supplemented with 0.025 mg/mL Gentamycin (G-1264, Sigma-Aldrich, St. Louis, MO, USA) and 3 mg/mL bovine serum albumin (BSA) (#A-6003, Sigma-Aldrich). The COCs were then matured in TCM199 media (#11150-059, Gibco) containing 0.1% polyvinyl alcohol (PVA) (w/v), 3.05 mM D-glucose, 0.91 mM sodium pyruvate, 75 µg/mL penicillin G, 50 µg/mL streptomycin, 0.57 mM cysteine, 10 ng/mL epidermal growth factor, 0.5 µg/mL luteinizing hormone, 0.5 µg/mL follicle-stimulating hormone, and 10% (v/v) porcine follicular fluid for 22 hours, this was followed by an additional 22 hours in the same medium but without the luteinizing and follicle-stimulating hormones, at 38.5°C under 5% CO2. After 44 hours of in vitro maturation (IVM), cumulus cells were removed by gentle pipetting following a 5-minute treatment with 0.1% hyaluronidase. Matured oocytes displaying the first polar body were enucleated by aspirating the polar body and surrounding cytoplasm containing metaphase II chromosomes using a beveled pipette (16–18 µm in diameter). Transgenic cells were then introduced into the perivitelline space of these enucleated oocytes. The oocytes along with transgenic cells were placed between 0.2-mm diameter electrodes in a fusion chamber filled with 0.3 M mannitol solution, containing 0.1 mM MgSO4, 1.0 mM CaCl2, and 0.5 mM HEPES. To induce fusion, two direct current (DC) pulses of 1.5 kV/cm were applied for 30 µs using a cell fusion generator (Nepa Gene, Ichikawa, Japan). After one hour of incubation in porcine zygote medium (PZM)-3 [43] with 0.3% (w/v) BSA, reconstructed embryos were surgically transferred into both oviducts of the surrogates on the same day or 1 day after estrus was observed.
Embryo transfer was conducted according to a previously described protocol, with slight modifications [42]. Surrogate sows aged 6–8 months and weighing between 120 and 150 kg were used. To sedate the surrogates, a solution (0.7 mg/kg) containing 10 mg/mL Alfaxalone (Alfaxan, Careside, Seongnam, Korea, Alfaxan) and 1 mg/mL dexmedetomidine (Dormitor, OrionPharma, Espoo, Finland) at a 9:1 ratio was administered through the auricular vein. To induce anesthesia in the pigs, isoflurane (35–40 mL/30 min/animal) was then administered via an anesthesia ventilator during the embryo transfer surgery. During anesthesia, a 5-cm midline incision was made to expose the uterus. After confirming the ovarian bleeding and follicular status, transgenic embryos were transplanted into the ampullary regions of both the left and right oviducts. After surgery, betadine was applied around the incision area. A single intramuscular injection of penicillin (400,000 IU/100 kg) was administered to prevent infection. Additionally, intramuscular injections of antibiotics (5 mL/100 kg, PPS, Daesung Microbiological Labs, Uiwang, Korea, ), methanpyrone (5 mL/sow, Novin-50, Bayer Korea, Seoul, Korea, ), and flumetasone (6–8 mL/150–200 kg, Fluson, Woogene B&G, Hwaseong, Korea, ) were administered to alleviate the pain associated with post-surgical inflammation. Once the surrogates had recovered from anesthesia, they were transferred to a recovery room for stabilization. Subsequent care and management of the surrogates were arranged in consultation with the veterinarian and assigned research personnel. The pregnancy status of the surrogates was monitored weekly using ultrasound starting four weeks after the embryo transfer. Cloned piglets were delivered via natural parturition. Genomic DNA extracted from the ear tissue or umbilical cord of the newborn piglets was used as the PCR template for genotyping.
To generate sgRNA constructs, directed against porcine ROSA26, CD163, ALK, and EML4 genes, the respective gRNA oligonucleotides (Supplemental Table 1) were synthesized and cloned with a 0.2-kb U6 promoter and an sgRNA scaffold sequence into EcoRI-HindIII- digested pUC57 vector using EcoRI and HindIII enzymes.
Genomic DNA, which was used as a template for the PCR, was extracted from the cells or tissues using a quick DNA Miniprep kit (Zymo Research, Irvine, CA, USA) following the instructions provided by the manufacturer; Platinum SuperFi II DNA Polymerase (Invitrogen, Waltham, MA, USA) was used according to the manufacturer’s instructions. Primers used for the PCR are listed in Supplemental Table 2.
To extract total RNAs, cells and tissues were directly lysed in TRIzol solution (Invitrogen) and then stored at −20°C until the RNA extraction procedure. RNA extraction was performed using a Direct-Zol RNA miniprep kit (Zymo Research) following the manufacturer’s protocol. RNA quality and concentration were assessed using a Nanodrop 1000 (Thermo Fisher Scientific). cDNA was synthesized using approximately 2000 ng of total RNA and the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA) as instructed by the manufacturer. The cDNA was stored at −20°C until use. The primers used in RT-PCR analyses are listed in Supplemental Table 2.
The pEFs, isolated from Cas9-expressing piglets, were cultured as previously described. Subsequently, 1 million cells were transfected with 2 μg of vectors expressing gRNAs designed for targeting genes using Nucleofector II/2b (Lonza, Basel, Switzerland) and the Ingenio electroporation kit (Mirus bio, Madison, WI, USA) following the protocol provided by the manufacturer. After culturing the cells for 48 hours, genomic DNA and RNA were extracted using the Direct-Zol DNA/RNA Miniprep kit (Zymo Research), and then used for further analyses.
The target gene was amplified using genomic DNA or cDNA and TaKaRa Ex Taq Polymerase (Takara Bio, San Jose, CA, USA) following the instructions provided by the manufacturer. For this purpose, PCR products were purified and inserted into a T-vector (Promega, Madison, WI, USA) as instructed by the manufacturer. The ligated mixtures were used to transform competent E. coli cells, and transformed colonies were selected through blue-white screening. Inserts present in the selected colonies were analyzed using Sanger sequencing. The primers used for TA cloning are listed in Supplemental Table 2.
To extract protein, tissues were homogenized in T-PER solution containing a protease and phosphatase inhibitor cocktail (#78441, Thermo Fisher Scientific). The protein concentration was measured using a BCA protein assay kit (#23227, Thermo Fisher Scientific). Next, 4X sample buffer (#B0007, Invitrogen) was added to the sample, and the samples were vortexed and incubated for 5 minutes at 70°C. Samples were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to a PVDF membrane. The membranes were blocked with 1X Tris-buffered saline with Tween 20 (TBST) containing 5% BSA and then incubated with anti-Cas9 (1:1,000, #MA1-201, Thermo Fisher) or anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:1,000, Cell Signaling Technologies Danver, MA, USA, #2118S) primary antibodies in 1X TBST with 5% BSA overnight at 4°C on a shaker. The membranes were then washed 3 times with 1X TBST and incubated with horseradish peroxidase (HRP)-linked anti-mouse for Cas9 (1:10,000, #7076P2, Cell Signaling Technologies) and HRP-linked anti-rabbit for GAPDH (#7074P2, Cell Signaling Technologies) in 1X TBST with 5% BSA for 1 hour at room temperature on a shaker. After washing 5 times with TBST, proteins on the membranes were visualized using an enhanced chemiluminescence detection kit (#34577, Thermo Fisher Scientific).
RESULTS
The ROSA26 locus is recognized for its capacity to enable abundant, ubiquitous, and stable expression of exogenous genes across various species. Based on prior research indicating the efficacy of this locus in gene expression systems [44], we selected the ROSA26 locus as the insertion site for the Cas9 gene to develop Cas9-expressing pigs. We then constructed a vector for insertion of the Cas9 gene into this locus (Fig. 1A). The vector also incorporated Cas9-tdTomato and Neomycin resistance genes flanked by CRE and FRT recombinase target sites, respectively. This design permitted conditional suppression of Cas9-tdTomato expression (Fig. 1B) and removal of the neomycin resistance gene (Fig. 1C) via CRE recombinase and Flippase, respectively, allowing for further regulatory control post-insertion.
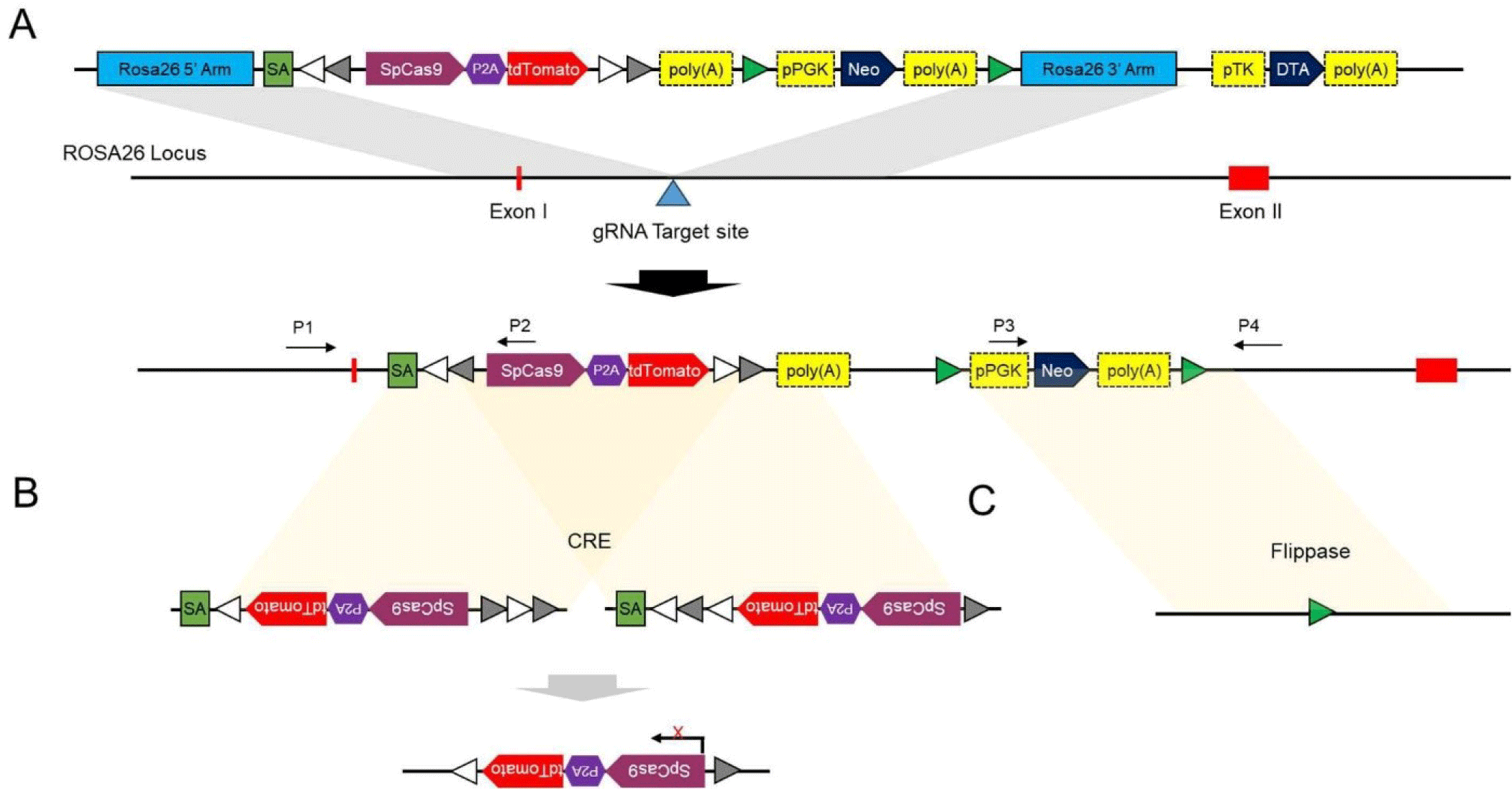
Cells expressing Cas9 were produced using pEFs isolated from 7-day-old male Nanchukmacdon piglets. The Cas9 gene cassette was linearized and introduced into the pEFs along with the Cas9 protein and gRNA complex targeting intron 1 of the ROSA26 locus. Cell colonies were selected using G418 (0.5 to 1.0 mg/mL) from day 3 to 21. Genotyping was performed via short-range PCR using primer pairs P1/P2 and P3/P4, which amplified the 5′ and 3′ junctions of the targeted allele, respectively. Additionally, long-range PCR spanning the 5′ and 3′ junctions was conducted using primers P1 and P4 (Supplemental Fig. 1). Among the 215 selected colonies, 11 (5.12%) were correctly targeted as confirmed by PCR. After losing one positive colony, further analysis was conducted on 10 colonies. Cas9 protein expression (Supplemental Fig. 2) and a normal karyotype (Supplemental Fig. 3) were detected in eight targeted colonies each. Ultimately, seven colonies exhibited both Cas9 protein expression and a normal karyotype.
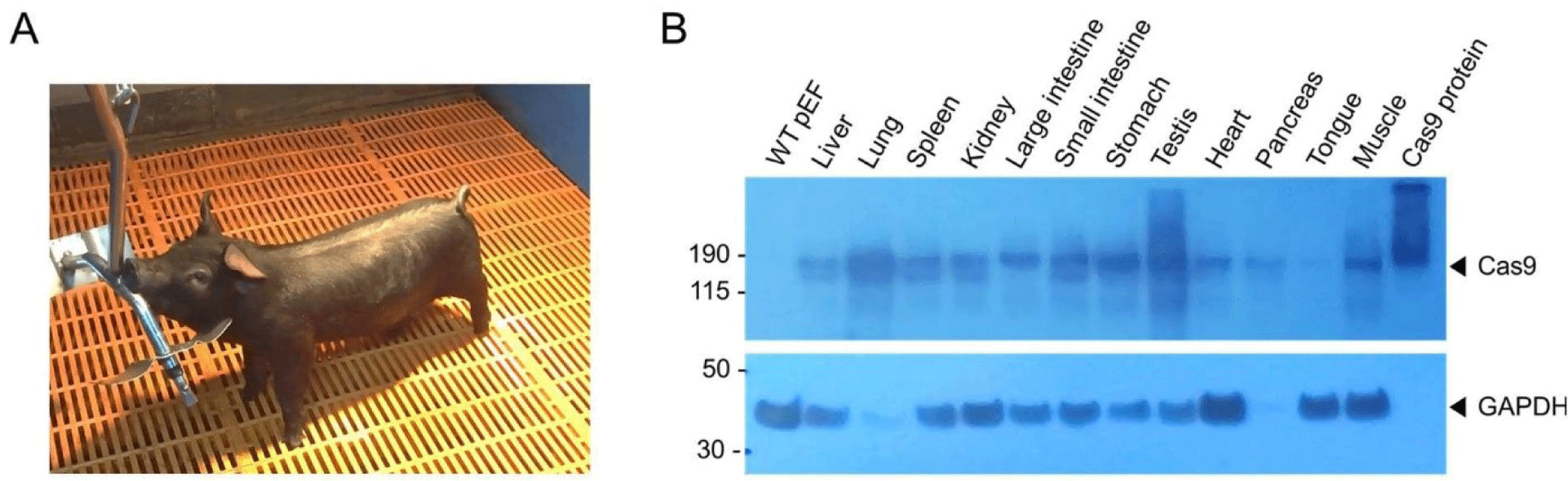
To generate Cas9-expressing pigs, we performed SCNT with Cas9-expressing pEFs as donor cells. we selected two Cas9 gene transgenic pEF colonies, designated as #88 and #164. Approximately 4,200 cloned embryos were derived from these donor cells and subsequently transferred into 14 surrogate sows. Thirteen cloned male piglets were delivered full-term after a gestation period of 117–120 days. Of these piglets, 11 died shortly after birth, and one succumbed after 10 days; and only one Cas9 heterozygous male piglet survived (Fig. 2A). We performed PCR across the 5′ and 3′ junctions of the targeted allele, which confirmed integration of the Cas9 gene cassette at the ROSA26 locus in pEFs from 12 of 13 piglets (Supplemental Fig. 4). Moreover, all pEFs derived from the piglets expressed the tdTomato protein, except for sample #12, which was lost because of contamination (Supplemental Fig. 5). Additionally, expression of the Cas9 protein was detected in various tissues, including the liver, heart, lung, spleen, kidney, testis, tongue, muscle, and both the large and small intestines, as well as the stomach, but not in the wild-type pEFs (Fig. 2B). Subsequently, to evaluate whether the surviving male piglet was fertile, we conducted artificial insemination (AI) using sperm derived from this pig. AI was performed on two wild-type female pigs, which resulted in the birth of twelve piglets (Supplemental Fig. 6). Of these, eight piglets were wild-type, while four carried the Cas9 gene. These findings suggest that the Cas9-expressing male pig was fertile and that the production of offspring through AI proceeded normally; however, the production efficiency of SCNT was low.
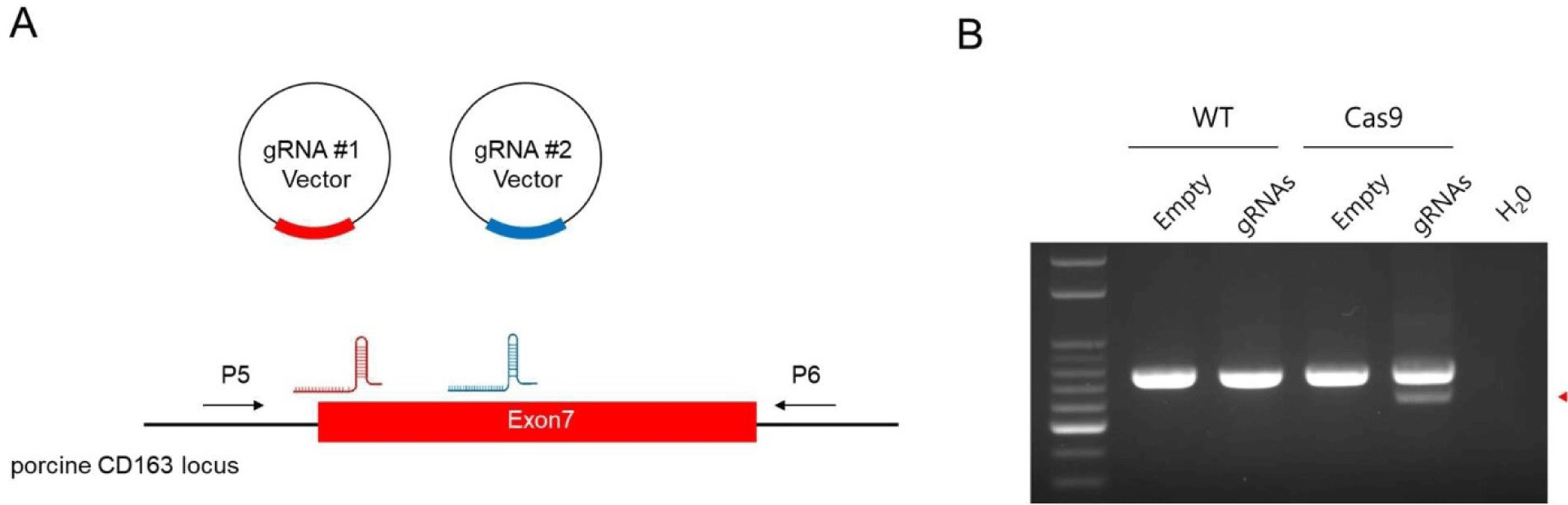
To assess the functionality of the Cas9 protein expressed in pigs, we targeted the CD163 gene, which is crucial for the entry of porcine reproductive and respiratory syndrome (PRRS) virus into cells. Prior studies have shown that domain 5, encoded by exon 7 in this gene, is vital for virus binding, and that pigs with deletions in this region exhibit resistance to PRRS virus (PRRSV) [9,45]. We constructed a vector expressing two gRNAs specific to exon 7 of the CD163 gene and introduced it into both wild-type and Cas9-expressing cells (Fig. 3A). Subsequent PCR analysis confirmed DNA mutations at the targeted sites within the CD163 gene (Fig. 3B). Sanger sequencing of the PCR products validated the presence of deletions and point mutations at the gRNA target sites, demonstrating the high efficiency of Cas9 activity in this cellular model (Supplemental Fig. 7)
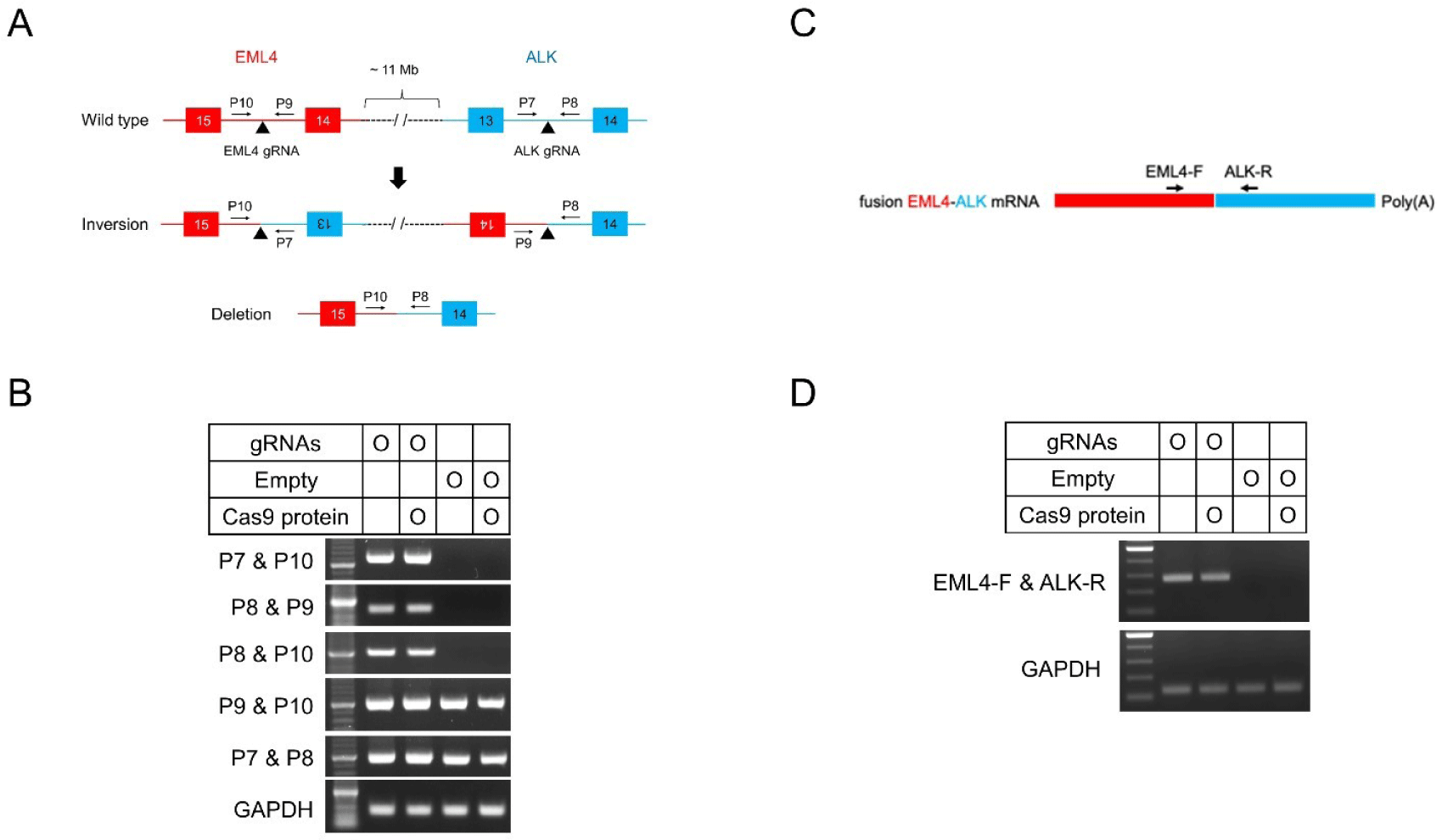
Pigs show chromosomal rearrangements between the genes encoding echinoderm microtubule-associated protein-like 4 (EML4) and anaplastic lymphoma kinase (ALK) [36]. We investigated the induction of rearrangements in Cas9-expressing pigs. Two sgRNA-expressing vectors were generated, one targeting EML4 and the other targeting ALK, which were simultaneously introduced into Cas9-expressing pEFs (Fig. 4A). Genomic DNA PCR performed 2 days post-transfection, showed that inversions and large deletions occurred in cells transfected with both sgRNA-expressing vectors but not in cells transfected with empty vectors (Fig. 4B). Moreover, chromosomal rearrangements can generate an in-frame fusion mRNA of EML4-ALK, comprising exons 1–14 of EML4 and exons 14–23 of ALK [46]. We investigated the occurrence of in-frame fusion (Fig. 4C). The RT-PCR and Sanger sequencing analyses revealed that in-frame fusion between the mRNA of EML4 and ALK occurred as a result of chromosomal inversion (Fig. 4D and Supplemental Fig. 8). These results confirm that the development of Cas9 transgenic pigs allowed the generation of indel mutations as well as chromosomal rearrangements, such as inversions and large fragment deletions mediated by sgRNAs.
DISCUSSION
GE pigs can be effectively used in both agriculture and biomedicine [47]. However, generating GE pigs with the appropriate phenotype for specific purposes is inefficient, laborious, and time-consuming owing to the variability in gene candidate functions between cellular and organismal levels. Although the CRISPR-Cas9 technique has increased the efficiency of modifying target genes, inefficiencies remain in other processes such as SCNT, embryo transfer, and validation of the suitability for intended purposes of the GE pigs produced. To overcome these challenges, other researchers have attempted to modify target genes in vivo by directly introducing the Cas9 protein and sgRNA [20–28,48,49]. However, this method has the disadvantage of reduced gene editing efficiency owing to the size of the Cas9 gene and the immune response of the animal itself [50]. To overcome this problem, several animals have been developed to inducibly and constitutively express the Cas9 protein [32,36–39,51,52]. Here, we successfully generated a fertile Cas9-expressing male pig and confirmed in vitro gene-editing with primary cells derived from this pig by simply introducing sgRNA, suggesting that the function of gene candidates could be evaluated in vivo.
To generate a Cas9-expressing pig, we chose the ROSA26 locus, renowned for its robustness in gene expression across various species [53,54]. The ROSA26 locus was specifically chosen owing to its proven efficacy in enabling stable and ubiquitous expression of exogenous genes in genetically engineered organisms, which is essential for consistent gene expression without silencing [44]. Indeed, we could obtain transgenic colonies stably expressing Cas9 protein with a normal karyotype, suggesting that the ROSA26 locus is an appropriate site for exogenous gene expression in pigs as well. Further, analysis with tissues from deceased Cas9-expressing piglets showed Cas9 protein expression across multiple tissues, confirming that the gene was correctly expressed and functional within the animal. In addition, AI with sperms from the pig successfully generated offspring, indicating that the pig was fertile.
Unlike previous studies, which generated Cre-dependent or Doxycycline-inducible Cas9-expressing pigs [36,39], our pig ubiquitously expressed a single copy of SpCas9 under the control of the endogenous ROSA26 promoter. This approach eliminated the need for recombination or induction events to activate Cas9 protein expression, which should enhance editing efficiency. Animals that ubiquitously express Cas9, including mice and pigs, have been reported to be healthy and fertile [32,38]. Despite the successful generation of transgenic cells, we obtained only one live Cas9-expressing male pig from 14 surrogates and 4,200 transgenic eggs via SCNT. However, AI using its sperm in two wildtype sows produced four Cas9-expressing piglets (~33%; 4 out of 12). This relatively lower production rate than expected (50%) might be attributed to the toxic effects associated with the ubiquitous and constitutive expression of the Cas9 protein. Studies have demonstrated that constant Cas9 activity can lead to DNA damage [55], chromosome instability [56], immune response [57], embryo lethality [38], enrichment of p53 mutants, and potential carcinogenic effects [58–60]. Therefore, future studies should focus on reducing the toxicity associated with ubiquitous and constitutive Cas9 expression by developing tissue-specific promoters or inducible systems to control Cas9 protein expression, as reported previously [39,46]. Additionally, as reported by Zhang et al. [61], developing engineered Cas9 variants with lower activity in animals could mitigate off-target effects and reduce cellular stress.
To access the functionality of Cas9 protein expressed in our pig, we chose CD163 as the target gene, which is known to be related to the invasion of PRRSV. PRRS is one of the most economically significant diseases affecting pig farming, which results in reproductive failures, including stillbirth, abortion, and premature farrowing [62]. After identification of PRRSV, numerous groups have developed protective approaches against PRRSV infection. Pigs carrying null alleles of the CD163 gene, which encodes a membrane receptor for the virus, have been developed, demonstrated resistance to PRRSV infection [9,63,]. The introduction of two synthetic CD163 gene-targeting sgRNAs into Cas9-expressing fibroblasts induced deletion of the gene, demonstrating the functionality of the Cas9 protein expressed in the pig.
To further investigate functionality, we selected EML4 and ALK considering the frequent chromosomal rearrangements between these genes in solid cancers in humans [62]; similar rearrangements have also been reported in pigs [36, 39]. Predictably, our Cas9-expressing pigs exhibited chromosomal rearrangements, including inversions and large deletions between EML4 and ALK, along with in-frame fusion mRNA transcripts of EML4 and ALK. The data suggest the functionality of the Cas9 protein in pigs and that Cas9-expressing pigs, along with their tissues and cells, can be considered effective tools for studying human diseases such as cardiovascular, neurodegenerative, metabolic, and other genetic diseases [64].
In conclusion, we successfully generated a Cas9-expressing porcine model and demonstrated efficient genome editing of different target genes using primary fibroblasts derived from pigs through a single delivery of engineered vectors encoding sgRNAs, thus confirming Cas9 functionality in the pigs. However, future studies are needed to determine whether gene editing can occur in vivo and to develop methods for increasing the production rate of Cas9-expressing pigs. We are confident that Cas9 transgenic pigs provide a powerful platform for in vivo and in vitro genome editing in agricultural and biomedical research.